3.8(Q2)
CiteScore
CiteScore
27
h-index
h-index
Document Type : Original Research Article
Authors
1 Department of Chemistry, University College of Science, Osmania University, Hyderabad, 500007, Telangana, India
2 Geethanjali College of Engineering and Technology, Cheeryal, Keesara, Medchal, 501301 Telangana, India
3 Department of Sciences and Humanities, Matrusri Engineering College, Saidabad, Hyderabad, 500059, Telangana, India
Graphical Abstract
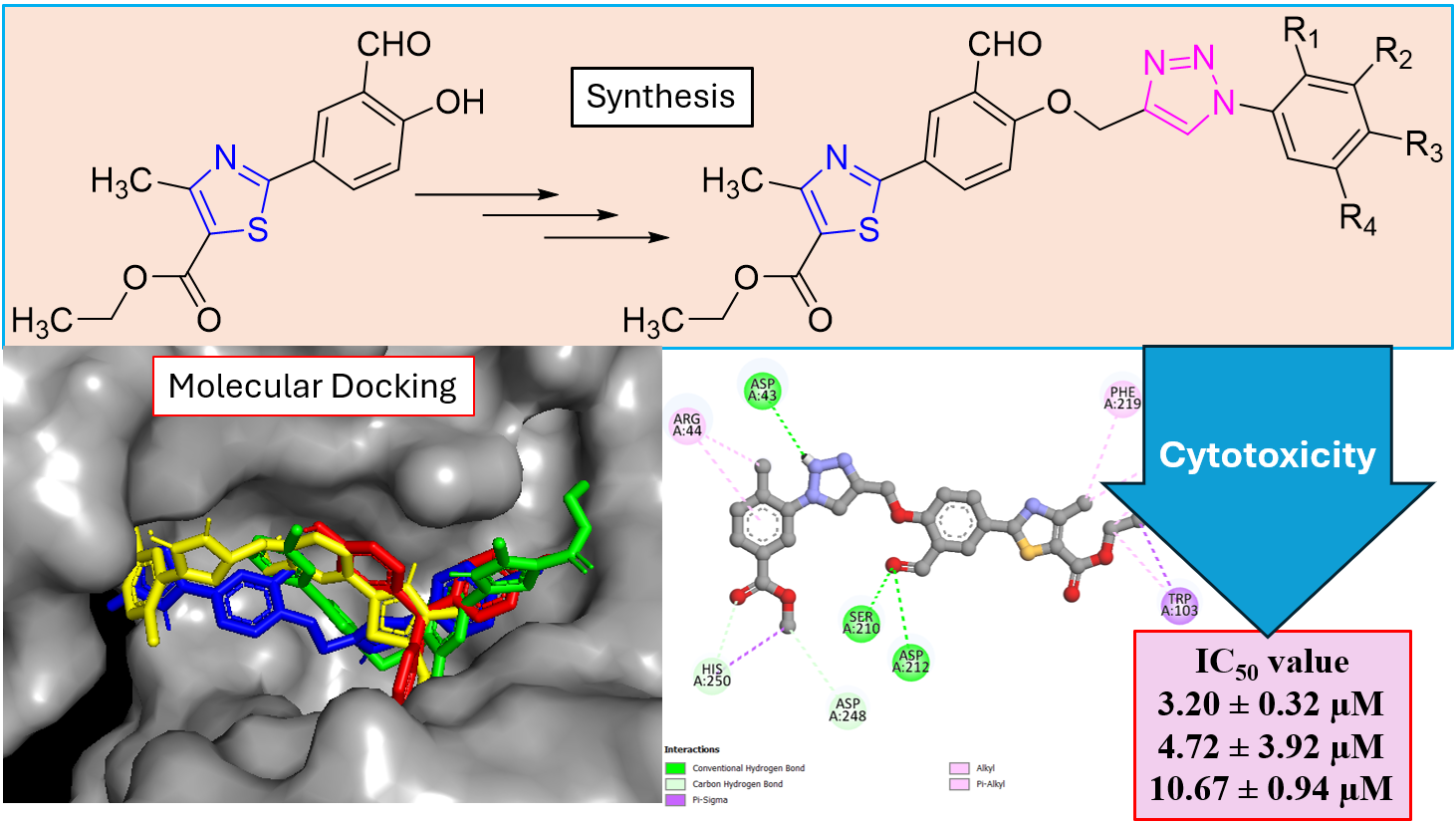
Keywords
Subjects
Introduction
Cancer is a disease characterized by aberrant, uncontrolled cell cycle progression and accelerated proliferation of normal cells. Cancer is the second biggest cause of mortality globally, with a rising incidence of new cases year, surpassed only by cardiovascular disorders. The incidence of cancer-related death is escalating at an unprecedented rate in numerous developing nations [1-3]. Despite significant advancements in cancer treatment over the past decade and the identification of numerous new chemotherapeutics, these chemicals exhibit considerable cytotoxicity and may lead to resistance, thereby diminishing medication efficacy. This lethal disease remains a dramatic challenge and is a critical topic for investigation [4]. Consequently, the design and development of novel cancer therapies continue to provide a substantial and formidable challenge for medicinal chemists globally [5]. Thiazoles and their derivatives represent a class of heterocyclic compounds distinguished by their significant biological properties, earning recognition in medicinal chemistry as potential therapeutic agents of considerable promise [6]. Consequently, spontaneous techniques are employed in medicinal chemistry to synthesize various classes of thiazole compounds that show significant potential in combating cancer cells [7]. Compounds containing thiazole are presently incorporated in various clinically approved anticancer medications. Viz. Tiazofurin inhibits IMP dehydrogenase and is utilized as an antineoplastic drug specifically for the treatment of Philadelphia chromosome (Ph)-positive leukemias [8]. Dasatinib is an orally delivered multi-kinase inhibitor, while Dabrafenib is an orally supplied multikinase inhibitor that selectively targets the B-RAF enzyme. It serves as an antineoplastic and an anticoronaviral agent [9,10]. Ixabepilone, licensed by the FDA, is utilized for the treatment of refractory aggressive metastatic or locally advanced breast cancer [11]. Fatostatin inhibits the proliferation of endometrial cancer in endometrial carcinoma cells inside a xenograft model via modulating lipid metabolism [12] (Scheme 1). The thiazole ring performs as a multifaceted scaffold in drug conception, with its derivatives exhibiting a range of pharmacological applications across multiple disease domains, rendering them appealing targets for medicinal chemists and pharmaceutical researchers [13-16].
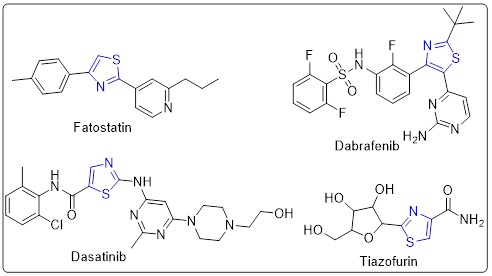
Scheme 1. Anticancer drugs containing thiazole ring
The molecular framework of 1,2,3-triazole displays unique physicochemical properties, including an amide bond, resemblance to amide bioisosteres, low basicity, stable metabolism, and the ability to act as a hydrogen-bond acceptor [17]. The properties facilitate the binding of bio-molecular targets and improve solubility [18,19]. It has been demonstrated that medications based on 1,2,3-triazole, such as Mubritinib and Carboxyamido-triazole, had powerful anti-cancer capabilities [20-22]. Based on these breakthroughs, we were inspired to create a new series of thiazole-based 1,2,3-triazole hybrid molecules. We used thiazole-derived alcohol as the primary precursor, and we evaluated their anticancer activity in vitro against U87 glioblastoma cell lines. In addition, we carried out a molecular docking study in order to gain a better understanding of their binding interactions.
Experimental
Without any additional purification, each and every one of the reagents and solvents that were acquired from Sigma-Aldrich were utilized immediately. The digital melting point device manufactured by Labtronics in Panchkula, India, was used to record melting points in degrees Celsius. Using the ATR technique, IR spectra were recorded on the Bruker-27 instrument. Using TMS as an internal reference in CDCl3 solvent, the 1H-NMR and 13C-NMR spectra were acquired. These spectra were taken using a JEOL JNM-ECZ500R/S1 instrument at 400 MHz and 100 MHz, respectively. Investigations using Mass spectrometry were conducted out on a Jeol SX-102 spectrometer, which was manufactured in Tokyo, Japan. To predict the reaction progression, TLC plates and aluminium sheets that had been pre-coated with silica gel 60 F254 were utilized.
Procedure for the synthesis of Ethyl 2-(3-formyl-4-(prop-2-yn-1-yloxy) phenyl)-4-methylthiazole-5-carboxylate (3)
Propargyl bromide (2) (1.4 g, 11.82 mmol) was gradually introduced to a stirring mixture of ethyl 2-(3-formyl-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate (1) (1 g) and anhydrous K2CO3 (1.48 g, 10.75 mmol) in dry DMF (15 ml). The mixture was then stirred at room temperature for 5 hours. Upon the reaction completion, as checked by TLC, the resultant mixture was poured into water and extracted alongside ethyl acetate (3×15 ml). The organic layer was washed with water (3×25 ml), and then dried, filtered, and concentrated. The crude product underwent purification through column chromatography with a pet-ether: EtOAc mixture to yield the pure product. Yield: 75%; m.p.: 126 ºC. IR (ATR, νmax, cm-1): 1685 (CHO) and 2127 (C≡CH); 1H-NMR (500 MHz, CDCl3) δ 10.49 (s, 1H), 8.39 (s, 1H), 8.24 (d, J = 8.7 Hz, 1H), 7.47 (s, 1H), 4.91 (s, 2H), 4.35 (q, J = 7.0 Hz, 2H), 2.77 (s, 3H), 2.61 (s, 1H), and 1.39 (t, J = 7.0 Hz, 3H); 13C-NMR (126 MHz, CDCl3): ) δ 188.6, 168.1, 162.3, 161.2, 161.1, 133.6, 127.3, 127.0, 125.6, 121.9, 113.8, 77.5, 76.8, 61.4, 56.7, 17.6, and 14.4. ESI-MS: m/z 330.21[M+H] +.
General procedure for the synthesis of ethyl 2-(3-formyl-4-((1-phenyl-1H-1,2,3-triazol-4-yl) methoxy) phenyl)-4-methylthiazole-5-carboxylate (5a-k)
Various individual substituted phenyl azides (4a-k) (0.25 g) and ethyl 2-(3-formyl-4-(prop-2-yn-1-yloxy) phenyl)-4-methylthiazole-5-carboxylate (3) (0.141 g) were dissolved in DMF (5 ml) solvent. To this, sodium ascorbate (0.3 mmol, 300 µL of a freshly made 1 M solution in water) was added, followed by copper (II) sulfate pentahydrate (7.5 mg, 0.03 mmol, in 100 µL of water). The uniform mixture was agitated continuously overnight, resulting in clarification, and TLC analysis confirmed the full utilization of the reactants. The reaction mixture was diluted with 50 cc of water, chilled on ice, and the precipitate was recovered by filtration. Following the washing the precipitate with cold water (2×25 ml), it was dried under vacuum to obtain a pure product in substantial quantities.
Spectra
Ethyl 2-(3-formyl-4-((1-phenyl-1H-1,2,3-triazol-4-yl) methoxy) phenyl)-4-methylthiazole-5-carboxylate (5a)
Yield: 84%; m.p.: 200 ºC. IR (ATR, νmax, cm-1): 1686 (CO) and 1598 (CHO); 1H-NMR (CDCl3, 400 MHz): δ 10.49 (s, 1H), 8.38 (d, J = 2.0 Hz, 1H), 8.23 (dd, J = 8.7, 1.9 Hz, 1H), 8.14 (s, 1H), 7.76 (d, J = 7.9 Hz, 2H), 7.51 (dt, J = 15.6, 7.3 Hz, 3H), 7.34 (d, J = 8.8 Hz, 1H), 5.51 (s, 2H), 4.35 (q, J = 7.1 Hz, 2H), 2.77 (s, 3H), and 1.39 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 188.6, 168.1, 162.2, 161.8, 161.1, 136.8, 133.8, 129.9, 129.1, 127.9, 127.7, 126.6, 125.2, 121.8, 121.3, 120.6, 113.7, 71.8, 61.3, 17.5, and 14.3; ESI-MS: m/z 449.28 [M+H] +.
Ethyl 2-(4-((1-(4-fluorophenyl)-1H-1,2,3-triazol-4-yl) methoxy)-3-formylphenyl)-4-methylthiazole-5-carboxylate (5b)
Yield: 82%; m.p.: 180 ºC. IR (ATR, νmax, cm-1): 1686 (CO) and 1598 (CHO); 1H-NMR (CDCl3, 400 MHz): δ 10.49 (s, 1H), 8.39 (s, 2H), 8.24 (d, J = 8.1 Hz, 2H), 7.73 (s, 1H), 7.34 (d, J = 8.6 Hz, 1H), 7.22 (d, J = 8.7 Hz, 2H), 4.91 (s, 2H), 4.36 (q, J = 7.2 Hz, 2H), 2.77 (s, 3H), and 1.39 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 188.5, 168.1, 163.2, 162.2, 161.7, 161.1, 144.9, 133.5, 133.0, 131.1, 127.7, 127.3, 126.8, 122.7, 117.0, 116.8, 115.2, 73.3, 61.3, 17.5, and 14.3; ESI-MS: m/z 467.27 [M+H] +.
Ethyl 2-(3-formyl-4-((1-(4-iodophenyl)-1H-1,2,3-triazol-4-yl) methoxy) phenyl)-4-methylthiazole-5-carboxylate (5c):
Yield: 86%; m.p.: 186 ºC. IR (ATR, νmax, cm-1): 1686 (CO) and 1600 (CHO); 1H-NMR (CDCl3, 400 MHz): δ 10.48 (s, 1H), 8.38 (dd, J = 4.0, 2.4 Hz, 1H), 8.32-8.15 (m, 1H), 8.13 (s, 1H), 7.88 (d, J = 8.8 Hz, 2H), 7.52 (d, J = 8.8 Hz, 2H), 7.32 (d, J = 8.8 Hz, 1H), 5.49 (s, 2H), 4.35 (q, J = 7.1 Hz, 2H), 2.77 (s, 3H), and 1.39 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 188.5, 168.0, 162.1, 161.7, 161.0, 143.8, 139.0, 136.4, 133.8, 133.5, 127.6, 127.3, 126.8, 122.1, 121.0, 113.6, 94.1, 62.6, 61.3, 17.5, and 14.3; ESI-MS: m/z 575.22 [M+H] +.
Ethyl 2-(4-((1-(4-bromophenyl)-1H-1,2,3-triazol-4-yl) methoxy)-3-formylphenyl)-4-methylthiazole-5-carboxylate (5d):
Yield: 75%; m.p.: 196 ºC. IR (ATR, νmax, cm-1): 1686 (CO) and 1600 (CHO); 1H-NMR (CDCl3, 400 MHz): δ 10.48 (s, 1H), 8.38 (dd, J = 4.0, 2.4 Hz, 1H), 8.32-8.15 (m, 1H), 8.13 (s, 1H), 7.88 (d, J = 8.8 Hz, 2H), 7.52 (d, J = 8.8 Hz, 2H), 7.32 (d, J = 8.8 Hz, 1H), 5.49 (s, 2H), 4.35 (q, J = 7.1 Hz, 2H), 2.77 (s, 3H), and 1.39 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 188.6, 168.0, 162.2, 161.7, 161.1, 145.2, 135.7, 133.9, 133.1, 131.7, 127.7, 126.8, 125.3, 124.9, 122.9, 122.1, 113.6, 62.7, 61.3, 17.5, and 14.4.; ESI-MS: m/z 527.21 [M+H] +.
Ethyl 2-(3-formyl-4-((1-(p-tolyl)-1H-1,2,3-triazol-4-yl) methoxy) phenyl)-4 methyl thiazole-5-carboxylate (5e)
Yield: 70%; m.p.: 198 ºC. IR (ATR, νmax, cm-1): 1686 (CO) and 1686 (CHO); 1H-NMR (CDCl3, 400 MHz): δ 10.48 (s, 1H), 8.37 (d, J = 1.8 Hz, 1H), 8.26-8.17 (m, 1H), 8.11 (s, 1H), 7.62 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 7.7 Hz, 3H), 5.49 (s, 2H), 4.35 (q, J = 7.1 Hz, 2H), 2.76 (s, 3H), 2.43 (s, 3H), and 1.39 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 188.6, 168.1, 162.2, 161.9, 161.0, 144.6, 139.3, 134.5, 133.8, 130.3, 127.5, 126.7, 126.1, 125.2, 121.8, 120.5, 111.7, 62.7, 61.3, 21.1, 17.5, and 14.3; ESI-MS: m/z 463.31 [M+H] +.
Ethyl 2-(4-((1-(3-chlorophenyl)-1H-1,2,3-triazol-4-yl) methoxy)-3-formylphenyl)-4-methylthiazole-5-carboxylate (5f)
Yield: 85%; m.p.: 196 ºC. IR (ATR, νmax, cm-1): 1717 (CO) and 1685(CHO); 1H-NMR (CDCl3, 400 MHz): δ 10.49 (s, 1H), 8.39 (d, J = 2.2 Hz, 1H), 8.24 (dd, J = 9.1, 2.2 Hz, 1H), 8.14 (s, 1H), 7.82 (s, 1H), 7.67 (d, J = 7.7 Hz, 1H), 7.53-7.43 (m, 2H), 7.33 (d, J = 8.8 Hz, 1H), 5.51 (s, 2H), 4.36 (q, J = 7.1 Hz, 2H), 2.77 (s, 3H), and 1.39 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 188.5, 165.8, 162.5, 161.9, 161.4, 140.5, 134.7, 133.9, 131.3, 131.0, 129.3, 126.9, 125.3, 122.3, 121.5, 120.9, 118.6, 115.8, 113.6, 71.2, 61.3, 17.5, and 14.3; ESI-MS: m/z 483.24 [M+H] +.
Ethyl 2-(4-((1-(3,5-dimethoxyphenyl)-1H-1,2,3-triazol-4-yl) methoxy)-3-formylphenyl)-4-methylthiazole-5-carboxylate (5g)
Yield: 85%; m.p.: 160 ºC. IR (ATR, νmax, cm-1): 1698 (CO) and 1679(CHO); 1H-NMR (CDCl3, 400 MHz): δ 10.49 (s, 1H), 8.38 (d, J = 2.3 Hz, 1H), 8.22 (dd, J = 8.7, 2.2 Hz, 1H), 8.11 (s, 1H), 7.33 (d, J = 8.8 Hz, 1H), 6.90 (d, J = 2.1 Hz, 2H), 6.53 (t, J = 2.0 Hz, 1H), 5.49 (s, 2H), 4.35 (q, J = 7.1 Hz, 2H), 3.86 (s, 6H), 2.77 (s, 3H), and 1.39 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 188.9, 168.0, 162.2, 161.8, 161.6, 161.0, 143.4, 138.2, 133.8, 127.5, 126.7, 125.2, 121.8, 121.5, 113.6, 100.8, 99.1, 62.7, 61.3, 55.7, 17.5, and 14.3; ESI-MS: m/z 509.40 [M+H] +.
Ethyl2-(3-formyl-4-((1-(5-(methoxycarbonyl)-2-methylphenyl)-1H-1,2,3-triazol-4-yl) methoxy) phenyl)-4-methylthiazole-5-carboxylate (5h)
Yield: 85%; m.p.: 165 ºC. IR (ATR, νmax, cm-1): 1698 (CO) and 1679(CHO); 1H-NMR (CDCl3, 400 MHz): δ 10.49 (s, 1H), 8.39 (d, J = 2.2 Hz, 1H), 8.25 (dd, J = 8.7, 2.2 Hz, 1H), 8.14 – 8.08 (m, 1H), 8.03 (s, 1H), 7.95 (s, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.36 (d, J = 8.8 Hz, 1H), 5.53 (s, 2H), 4.36 (q, J = 7.1 Hz, 2H), 3.93 (s, 3H), 2.77 (s, 3H), 2.31 (s, 3H), and 1.39 (t, J = 7.1 Hz, 3H);13C-NMR (101 MHz, CDCl3): δ 188.5, 168.0, 165.6, 162.1, 161.8, 161.0, 139.1, 136.2, 133.8, 131.9, 131.0, 129.3, 127.5, 127.0, 126.7, 125.7, 125.2, 121.8, 118.8, 113.6, 62.7, and 61.3; ESI-MS: m/z 521.42 [M+H] +.
Ethyl 2-(4-((1-(4-acetylphenyl)-1H-1,2,3-triazol-4-yl) methoxy)-3-formylphenyl)-4-methylthiazole-5-carboxylate (5i)
Yield: 76%; m.p.: 150 ºC. IR (ATR, νmax, cm-1): 1714 (CO) and 1681 (CHO); 1H-NMR (400 MHz, CDCl3) δ 10.49 (s, 1H), 8.38 (d, J = 8.7 Hz, 1H), 8.23 (d, J = 7.2 Hz, 2H), 8.14 (d, J = 8.4 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.7 Hz, 1H), 5.52 (s, 2H), 4.35 (q, J = 7.1 Hz, 2H), 2.77 (s, 3H), 2.67 (s, 3H), and 1.39 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 196.5, 188.5, 168.0, 162.2, 161.7, 161.1, 139.8, 137.2, 133.9, 130.2, 127.7, 126.9, 125.3, 122.8, 121.9, 121.1, 120.2, 113.6, 62.6, 61.3, 26.7, 17.5, and 14.3. ESI-MS: m/z 491.35 [M+H] +.
Ethyl 2-(3-formyl-4-((1-(3-nitrophenyl)-1H-1,2,3-triazol-4-yl) methoxy) phenyl)-4-methylthiazole-5-carboxylate (5j)
Yield: 70%; m.p.: 175 ºC. IR (ATR, νmax, cm-1): 1714 (CO) and 1681 (CHO); 1H-NMR (400 MHz, CDCl3) δ 10.50 (s, 1H), 8.65 (s, 1H), 8.38 (d, J = 13.7 Hz, 2H), 8.25 (d, J = 11.0 Hz, 2H), 7.78 (d, J = 7.6 Hz, 1H), 7.53 (d, J = 7.7 Hz, 1H), 7.33 (d, J = 8.7 Hz, 1H), 5.53 (s, 2H), 4.36 (q, J = 7.2 Hz, 2H), 2.77 (s, 3H), and 1.39 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 188.5, 168.0, 162.1, 161.7, 161.0, 143.8, 139.0, 136.4, 133.8, 133.5, 127.6, 127.3, 126.8, 122.1, 121.0, 113.6, 94.1, 62.6, 61.3, 17.5, and 14.3; ESI-MS: m/z 494.27 [M+H] +.
Ethyl 2-(4-((1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl) methoxy)-3-formylphenyl)-4-methylthiazole-5-carboxylate (5k)
Yield: 72%; m.p.: 210 ºC. IR (ATR, νmax, cm-1): 1714 (CO) and 1681 (CHO); 1H-NMR (400 MHz, DMSO) δ 10.43 (s, 1H), 9.07 (s, 1H), 8.30 (d, J = 2.4 Hz, 1H), 8.00 (s, 1H), 7.97 (s, 1H), 7.70 (d, J = 8.9 Hz, 2H), 7.65 (d, J = 9.5 Hz, 1H), 5.57 (s, 2H), 4.31 (q, J = 7.1 Hz, 2H), 2.70 (s, 3H), and 1.31 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 188.7, 175.7, 162.3, 161.8, 161.3, 144.6, 143.6, 135.1, 134.0, 130.3, 129.5, 127.8, 125.4, 124.3, 122.0, 116.8, 113.8, 71.5, 61.4, 17.6, and 14.4; ESI-MS: m/z 483.23 [M+H] +.
MTT assay
Cell viability was evaluated by the MTT Assay with five concentrations of the compound examined in triplicate. Cells were trypsinized and analyzed using the trypan blue technique to assess cell viability in the suspension. Cells were counted with a hemocytometer and injected at a density of 5.0×10-3 cells per well in 100 μl of culture media in a 96-well plate, subsequently incubated overnight at 37 °C. After incubation, the old media were discarded and 100 µl of fresh media was added with different quantities of the material into the specified wells of the 96-well plates. After 48 hours, the solution was eliminated and fresh medium was added containing MTT solution (0.5 mg/mL) to each well, and then incubated the plates at 37 °C for 3 hours. Following the incubation period, precipitates occurred as a result of the transformation of MTT salt into formazan crystals by cells with metabolically active mitochondria. The optical density of dissolved crystals in DMSO was measured at 570 nm with a microplate reader. The growth inhibition ratio was calculated using the following formula. The IC50 value was ascertained using the linear regression equation y = mx + c. In this context, y equals 50, whereas the values of m and c were obtained from the viability graph.
Molecular docking method
Using the Autodock Vina integrated PyRx tool [23-27], we conducted molecular docking studies against the crystal structure of thymidylate synthase (PDB ID: 6GYJ) obtained from the Protein Data Bank (www.rcsb.org). Protein was prepared by eliminating molecules of water and heteroatoms as well followed by the addition of polar hydrogens. Ligands have been drawn in ChemDraw Professional 16.0 using the MDL file format. The 3D grid box had been set up with dimensions of center_x = 95.3497133256, center_y = -13.8202751514, center_z = 39.5552748293, size_x = 24.0275270763, size_y = 24.5203069143, and size_z = 33.0607487873, with simulations conducted at an exhaustiveness of 8. The results were illustrated with Pymol and Biovia Discovery Studio.
Results and Discussion
Chemistry
Synthesis of ethyl 2-(3-formyl-4-((1-(substituted phenyl)-1H-1,2,3-triazol-4-yl) methoxy) phenyl)-4-methylthiazole-5-carboxylate derivatives (5a-k) depicted in Scheme 2. In the opening step, the precursor ethyl 2-(3-formyl-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate 1 was treated with propargyl bromide 2 in being there of potassium carbonate to get ethyl 2-(3-formyl-4-(prop-2-yn-1-yloxy) phenyl)-4-methylthiazole-5-carboxylate 3. It was proved by manifestation of two singlets at δ 4.91 ppm (2H) and δ 2.61 (1H) in 1H-NMR spectrum of compound 3 related to -CH2- and ≡CH protons of propargyl group, respectively. The carbonyl protons had seemed as a singlet at δ 10.49 ppm, the -CH3 group protons on thiazole ring appeared as a singlet at δ 2.77 ppm, the -CH3 and -CH2- protons of ethyl carboxylate group appeared as a triplet at δ 1.39 ppm and a quartet at δ 4.35 ppm, and three aromatic protons on phenyl appeared in downfield between δ 8.39 ppm to δ 7.47 ppm. In 13C-NMR spectrum of compound 3, the two signals appeared at δ 188.6 ppm and δ 168.1 ppm confirmed the carbonyl group carbons, the six aliphatic carbon signals corresponding to methyl, propargyl and ethyl groups appeared at δ 77.4, 76.8, 61.4, 56.7, 17.5, and 14.4 ppm. The mass spectrum of compound 3 recorded its m/z 330.21 [M+H] + peak. Finally, the terminal alkyne group of intermediate compound 3 was imperiled to copper catalyzed Huigen’s 1,3 dipolar cycloaddition click reaction with various substituted aryl azides 4a-k individually using catalytic quantities of copper sulphate pentahydrate and sodium ascorbate to get target compounds 5a-k. The characterization of compound 5a, depicted for example, in 1H-NMR spectrum, desertion of the signet at δ 2.61 ppm (≡CH) of propargyl group and appearance of a singlet at δ 8.14 ppm (triazole proton) authorized the formation of triazole ring. The -CH2- protons flumped between triazole and benzaldehyde group appeared as a singlet at δ 5.51 ppm. The signals of ethyl carboxylate and methyl group were retained in 1H- NMR spectrum. In addition, five protons integrated in aromatic region of 1H NMR spectrum confirmed the presence of new phenyl ring. In 13C-NMR spectrum, confirmed the disappearance of two carbon signals in aliphatic region corresponding to -C≡CH group. The two carbonyl carbon signals at δ 188.6 ppm and δ 168.1 ppm. The ESI-MS spectrum of compound 5a endorsed m/z 449.28 [M+H] + peak.
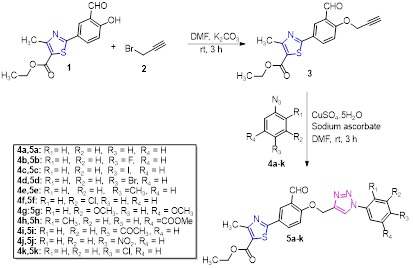
Scheme 2. Synthesis of ethyl 2-(3-formyl-4-((1-(substituted phenyl)-1H-1,2,3-triazol-4-yl) methoxy) phenyl)-4-methylthiazole-5-carboxylate derivatives (5a-k)
Anticancer activity
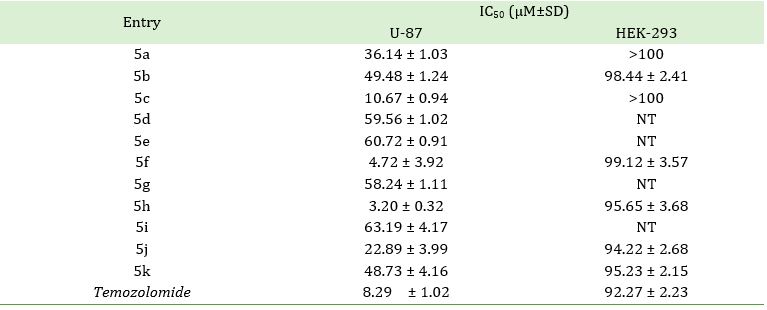
The newly synthesized compounds were screened for their in vitro anticancer activity against human glioblastoma cell lines (U87) engaging Temozolomide as standard reference and tested their cytotoxicity against normal human embryonic kidney cell cline (HEK-293), presented their IC50 value in Table 1. The activities of these compounds found to be good to moderate. Three compounds viz. 5c, 5f, and 5h had demonstrated potent activity in comparison to Temozolomide (IC50 = 8.29±1.02 µM). A structure-activity relationship analysis (SAR) was conducted to examine their activities concerning different substituents. Presence of an electron-donating methyl and an electron-withdrawing methyl carboxylate group on compound 5h displayed outstanding activity with an IC50 value of 3.20±0.32 µM. Placing an electron-withdrawing -Cl function in meta position of compound 5f earned slight change in activity with an IC50 value of 4.72±3.92 µM. Replacing para position of phenyl group with bulky -I function in compound 5c diminished activity with an IC50 value of 10.67 ± 0.94 µM. Replacement of -I function with acetyl group in compound 5j revised its activity to moderate with an IC50 value of 22.89 ± 3.99 µM. The rest of the functional group present on these compounds presented moderate activities. Furthermore, tested toxicity of best active compounds against normal HEK-293 cell lines, and not found any significant changes.
Computational screening against thymidylate synthase (TS)
Thymidylate synthase (TS) is an important enzyme involved in the production of 2′-deoxythymidine-5′-monophosphate, which is a necessary building block for DNA synthesis. Due to this reason, this enzyme is an essential focus in cancer treatment [28]. The active site pocket of mouse thymidylate synthase is comprised of amino acid residues that can be categorized as charged (Arg72, Asp212, Glu81, and Lys71), hydrophobic (Leu186, Trp103, Ile102, Cys189, Phe74, Val73, Phe219, Leu215, Tyr252, Tyr129, and Phe85), and polar (Gln208, Asn220, Ser210, His250, and His190) groups [29].
Table 1. IC50 value of target compounds 5a-k
The potent ligands 5c, 5f, and 5h were docked into the active site pocket of crystal structure of TS (PDB ID: 6GYJ) to get an insight into their binding efficacies. The docking procedure validated by redocking the co-crystalized ligand FGT which presented a binding affinity value of -8.1 kcal/mol (RMSD = 1.09 Å). Ligand 5c scored a binding energy value of -8.5 kcal/mol, the formyl group oxygen of this compound displayed a key interaction with amino acid site Asp212 of TS, the bond distance was found to be 1.99 Å, indicating a strong interaction between ligand and amino acid. The benzaldehyde and triazole ring established two π-sulfur interactions with amino acid site Cys189 of TS. A π-π stacked interaction instituted by phenyl ring linked to triazole with amino acid site Phe219 along with other hydrophobic interactions, as depicted in Figure 1. The ligan 5f, scored a binding affinity value of -8.8 kcal/mol, it displayed two key interactions with His190 and Asn220 of TS, the bond distance revealed to be 2.23 Å and 1.94 Å respectively, suggesting strong interactions. A π-π stacked interaction observed like ligand 5c, two π-sulfur interactions, one by triazole ring with Cys189 and other one with S atom of thiazole ring with Phe74 of TS (Figure 2). The most potent ligand 5h, presented a docking score of -9.2 kcal/mol, and there were three key interactions discovered with Asp43 (2.19 Å), Ser210 (2.13 Å) and Asp212 (2.55 Å) of TS signifying strong interactions along with other hydrophobic interactions (Figure 3). The reference ligand FGT showed H-bond interaction with Asn220, and hydrophobic interactions with Ile102, Trp103 (π-T shaped), His190, Leu215, and Phe219 (π-π stacked) of TS (Figure 4). The 3D dock pose image of these ligands in active site pocket of TS presented in Figure 5, reveal that these molecules could best fit into the cavity of thymidylate synthase for showing effective inhibition.
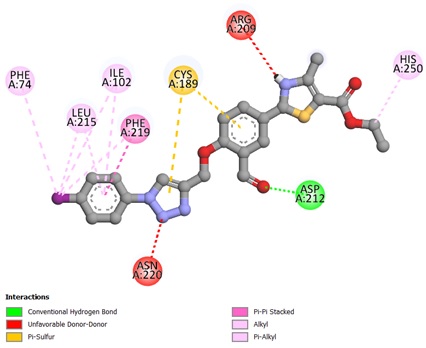
Figure 1. Binding interactions of ligand 5c against thymidylate synthase
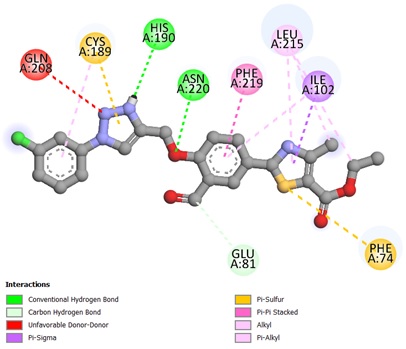
Figure 2. Binding interactions of compound 5f against thymidylate synthase
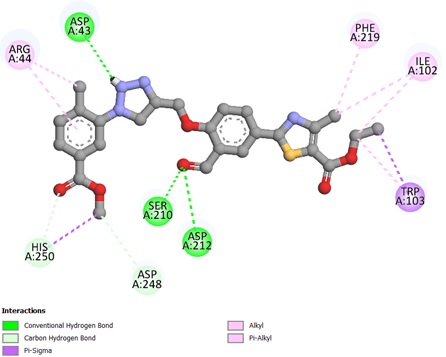
Figure 3. Binding interactions of ligand 5h against thymidylate synthase
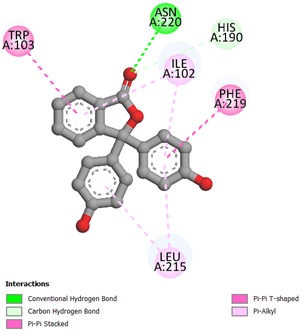
Figure 4. Binding interactions of FGT against thymidylate synthase
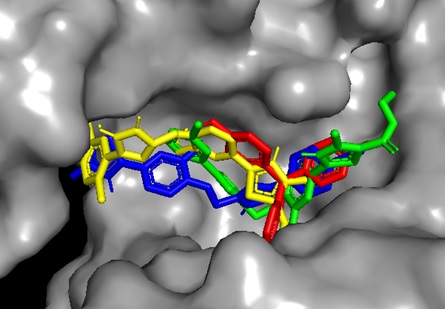
Figure 5. Dock pose of ligands 5c (blue), 5f (green), 5h (yellow), and FGT (red) in active site pocket of thymidylate synthase
Conclusion
The synthesis of a new series of triazoles linked to ethyl 2-(3-formyl-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate was reported, and their structures were characterized through the interpretation of 1H-NMR, 13C-NMR, and mass spectral data. The in vitro anticancer activity against human glioblastoma cell lines was evaluated for the three compounds 5c, 5f, and 5h. The results indicated potent activity, with IC50 values of 10.67±0.94 µM, 4.72±3.92 µM, and 3.20±0.32 µM, respectively, compared to Temozolomide. Conducted a computational study of their molecular interactions, yielding declaring docking scores and binding interactions, including hydrogen bonds, π-π stacking, and π-sulfur interactions. Therefore, these molecules necessitate further investigation for the invention of new chemotherapeutic options in the therapeutic management of glioblastoma cancer in the near future.
Acknowledgements
The authors would like to acknowledge the Head, Department of Chemistry for the provision of laboratory facilities at Osmania University, as well as CFRD Osmania University for their analytical support. In addition, appreciation is extended to the administration of Geetanjali College of Engineering and Technology and the Head of the F.E. Department for their support of the research efforts.
Disclosure Statement
The authors declare that there are no conflicts of interest pertaining to the publication of this article.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' Contributions
All authors contributed to data analysis, drafting, and revising of the article and agreed to be responsible for all the aspects of this work.
Orcid
Sateesh Kuna
https://orcid.org/0000-0001-5979-9740
Kotaiah Kandula
https://orcid.org/0000-0002-4601-8463
Nagaraju Myakala
https://orcid.org/0000-0002-2479-8921
Vishnu Thumma
https://orcid.org/0000-0001-6830-3671
Anagani Kanaka Durga Bhavani
https://orcid.org/0000-0002-5493-0663
How to cite this manuscript:
S. Kuna, K. Kandula, N. Myakala, V. Thumma, A.K.D. Bhavani. Synthesis of Novel Thiazole Based 1,2,3-Triazole Derivatives as Inhibitors of Glioblastoma Cancer Cells. Asian Journal of Green Chemistry, 9(2) 2025, 193-207.
DOI: https://doi.org/10.48309/AJGC.2025.491225.1603
URL: https://www.ajgreenchem.com/article_212130.html
PDF: https://www.ajgreenchem.com/article_212130_52b9c333185731408bee26741614528f.pdf
©2025 The author(s). This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit: http://creativecommons.org/licenses/by/4.0/
PUBLISHER NOTE
Sami Publishing Company remains neutral concerning jurisdictional claims in published maps and institutional affiliations.
CURRENT PUBLISHER
Sami Publishing Company

| Article View | 1,729 |
| PDF Download | 555 |