Document Type : Review Article
Author
Department of Chemistry, College of Science, University of Baghdad, Baghdad, Iraq
Abstract
Antibiotics called quinolones have a wide range of action, good oral absorption, and good bioavailability. Quinolones are capable of binding metallic ions and form complexes where they would operate as bidding, unidentating, and bridging ligands because of the chemical features within their nucleus, which include an O2 atom in the carbonyl group at place 4, a basic piperazinyl ring at site 7, and a carboxylic acid job at position 3. Quinolones hold onto the metal ions to form complexes that could function as bidentates, unidentates, or bridging ligands. Polymeric complexes in a solid state can be coordinated in various ways. Under extremely when exposed to acidic circumstances, quinolone molecules with a base end nucleus protonate and show up as cations in ionic complexes. The pharmacokinetics bioavailability and mode of action of these bactericidal drugs are all impacted by interactions with metal ions, which also have an impact on the solubility, pharmacokinetics, and bioavailability of quinolones. Many metal complexes were revealed to have antibacterial activity equivalent to or greater than the parent quinolones. Novel anticancer medications have come from the novel techniques for the formation of metal complexes of quinolones. The two primary areas of analytical applications of complex formation with metallic ions are metal I detection depending on complexation with quinolones and quinolone determination based on formation of complexes with metal ions.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
Quinolone antibiotics are a particular type of synthetic antibiotic that exhibits bactericidal activity, excellent oral absorption, and also a great bioavailability [1]. The 1960s saw the initial use of the family's first drug, nalidixic acid (1-ethyl-1,4-dihydro-7-methyl-4-oxo-1,8-naphthyridine-3-carboxylic acid in therapeutic settings, as displayed in Scheme 1.
Because of its restricted spectrum of action, the medicinal use of nalidixic acid has been limited [2]. Several alterations to the basic nucleus were made to extend the antibacterial range and enhancement pharmacokinetic quality, the two of which were deemed important: the F2 atom was added in location number six and the substitution of a piperazineone or more moiety N-heterocycle in the 7th site. In the 1980s, researchers found new 4-quinolones called fluoroquinolones. Quinolones was separated into 4 parts depending on the chemical composition of the basic nucleus (Scheme 2, Table 1). According to their antimicrobial range and pharmacokinetic characteristics, quinolones are divided into 4 productions [3].
Quinolones are antibacterial medications which prevent the transcription and replication of bacterial DNA, eventually bringing on cell damage. They prevent two enzyme DNA-gyrase (Topoisomerase II) and DNA topoisomerase IV from acting as antibacterial enzymes.

Scheme 1. Nalidixic acid

Scheme 2. The common configuration of 4-quinolones
Table 1. Quinolones are divided into generations according to antibacterial and pharmacokinetic characteristics range

The two DNA-gyrase subunits, GyrA and GyrB are responsible for introducing negatively transforms into DNA. By increasing the rate of daughter chromosome separation, four subunits make up DNA topoisomerase IV: The two components Par C and Par E subunits in charge of DNA decantation which enables cell division to produce two identical daughter cells [4]. Quinolones bind a combination of drugs, enzymes, and DNA that inhibit replication and progression is created when an enzyme-DNA complex is added to another enzyme [5]. In gram-negative bacteria, older quinolones have a greater inhibitory effect on DNA-gyrasein comparison with on topoisomerase IV, whereas in gram-positive bacterium, older quinolones have a greater inhibitory effect on topoisomerase IV. The two enzymes are similarly inhibited by more recent quinolones [6].
Background on quinolones
The only first quinolone medication, nalidixic acid, was identified the Sterling-Winthrop Research Organization began producing 1-alkyl-1,8-naphthyridines in 1962 as part of a series. A 2015 analysis of the history of quinolone antibiotics revealed that George Lesher, the creator of the 1962 book, had recorded the discovery of chloro-1-ethyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid. As a byproduct of chloroquine production in the late 1950s, with little antibacterial action, prompting additional research on analogs such as nalidixic acid [7]. Simultaneously, Imperial Chemical Industries (ICI) submitted patents for quinolone's antibacterial, containing a six-fluoroquinolone. Nalidixic acid is an antibacterial antibiotic with a restricted spectrum that was used to treat uncomplicated urinary tract infections (UTIs). Fluoric-quinolones, having a far greater field of action and superior pharmacokinetics than first-generation quinolones, significantly expanded the scope of the quinolone class in the 1970s and 1980s.
These fluoro quinolones, for instance, ciprofloxacin and ofloxacin, are effective compared with positively and negatively gram-stability infections, as well as mycobacterium tuberculosis, the causative agent of tuberculosis. For approximately 50 years, quinolones have been used as antibiotics because of their great efficacy, wideband of action, good absorption, simple formulations, high blood concentrations, and low incidence of side effects [8].
Development of quinolones
As a consequence of the creation of anti-malarial quinine molecules, only the first quinolone was found in the 1960s, nalidixic acid (technically a naphthyridone). The bacterial topoisomerase type II enzymes' activity was rapidly shown to be decreased, which prevented bacterial reproduction. The use of nalidixic acid as a treatment for gram-negative bacterial urinary tract infections received clinical approval in 1967 (UTIs) [9]. However, due to its limited range of effect, less the levels of serum attained, more inhibiting level needed, and some negative effecting so that only sometimes used. Better analogs weren't created until the 1980s, when the necessity for novel treatments for diarrhea and urinary tract infections brought on by resilient Shigella and Escherichia coli drew scientists' attention to enhance the effect and reduce the harmfulness of quinolones. Numerous investigations have been done on the relationships between the structure and action of quinolone antibiotics. Scheme 3 displays the basic configuration of the quinolones, from which two significant groups emerged: quinolones and naphthyridones, indicated by the 'X' place. In contrast to naphthyridones, which are characterized by the X location of the nitrogen atom, quinolones are specified by carbon X-positional atoms. Founded their range of activities, four groups exist for quinolones. Quinolones have been evolved from generation to generation by adding different substituents at various places on the pharmaceutical core to produce broader spectrum activity. The quinolone development route is summarized in Table 2.

Scheme 3. The fundamental structure of quinolone antibiotics. R1, R5, R6, R7, R8, and X are the six key places where changes can be made to increase the action of the drug. Naphthyridones are defined by X=N and quinolones by X=C
Table 2. Overview of the formation of quinolone antibiotic family



Activity development
Chief-generation quinolones only had an effect on Gram-negative bacteria, with the exceptional of Pseudomonas [10]. Soon after its clinical debut, nalidixic acid was shown to rapidly acquire resistance in a number of species, reducing its efficacy and spurring research to identify substitutes with enhanced qualities. The initial second-generation quinolone, flumequine, demonstrated how an important alteration, adding a fluorine atom at R6, may drastically alter the properties of the compound and expand the spectrum of action. First-generation quinolones only had an effect on Gram-negative bacteria, with instead of pseudomonas class. Soon after its clinical debut, nalidixic acid was demonstrated to rapidly cause improvement of resisting in a number of species, decreasing its efficiency, and encouraging the search for substitutes with enhanced qualities. Flumequine was also the primary second-generation quinolone showed how a crucial addition of a fluoride atoms at R6 position, may significantly expand the spectrum of action. The first-generation quinolones only had an effect on Gram-negative bacteria, with the excepting of Pseudomonas [10]. Soon after its clinical debut, nalidixic acid was shown to rapidly acquire resistance in a number of species, reducing its efficacy, and spurring research to identify substitutes with enhanced qualities. Adding a fluorine atom look at 6 sites to the first quinolone of the 2nd generation, flumequine demonstrated how this key alteration might greatly widen the range of action. Except for the most recent chemicals from the quarter generation, virtually all antibiotics of quinolone are now labeled as fluoroquinolones as a result of this change. Enoxacin, norfloxacin, and ciprofloxacin are further second-generation fluoroquinolones that prevent all Gram-negative organisms, which have containing kind of Pseudomonas [11]. A piperazine circle was added to the R7 place and a cyclopropyl molecule was added to the R1 location to further alter these medications in additional to the fluoro group position the cyclopropyl addition enhanced overall compound activity, whereas the R7 piperazine ring increased gram negative potency. Ciprofloxacin became the most effective drug in the initial compounds of the second generation as a result of this interaction, and it is currently the preferred therapy for pseudomonas aeruginosa. The second generation created analogs that were effective against a variation of gram-plus bacteria, containing mycoplasma pneumonia and chlamydia pneumonia, but not streptococcus pneumonia or staphylococcus aureus. One of the initial modifications that helped with the suppression of gram-positive organisms was the addition of an alkylation of piperazine crowd look at the R (seven), as demonstrated in ofloxacin [12]. The latter group's R8 location received a considerable boost in Gram-positive activity by receiving a -OCH3 substitution. Ofloxacin is still utilized in clinical therapy making it the most powerful of all the substances in the latter group (2b) since it contains all of the new substituents. The L-isomer of ofloxacin, a chiral molecule, is the only one that is active (levofloxacin). It was asserted that it is more effective than ciprofloxacin and has four times the activity of ofloxacin in treating certain strains. The quinolones reached their third generation with the development of fleroxacin. In this generation, pyrrolodinyl and alkylated piperazine groups were inserted in the R7 point, althoughamines, hydroxyl, and alkyl groups were added to the R5 place in the pharmacore. The cyclopropyl obtained at R1 and methoxy at R8 of the second generation were both unaffected. One of the additional substituents added to the third generation increased the anti-Gram-positive efficacy of the drug by placing a chlorine founded at the R8 site. In the present of the creation of fleroxacin, the quinolones have reached to the 3rd generational. Alkylated piperazine and pyrrolodinyl collections were added to the R7 position in this generation, as well as ammine, hydroxyl, and alkyl groups to the R5 station in the pharmacore. The second generation's cyclopropyllooking at R (one) and the methoxy group at R8 were both unaltered. A chlorine (Cl) which look at the R(8) site was one of the extra substituents included in the third generation, which was found to increase the drug's anti-Gram-positive activity. 8-methoxyquinolone exceeded all other medicines at this site in terms of activity and spectrum. Grepafloxacin and gatifloxacin are the two antibiotics that demonstrate the improvement the best; gatifloxacin (8-MeO) MIC90 significantly outperformed grepafloxacin (8-H) in this comparison (Table 3). These modifications improved the third generation's action against atypical bacteria and its Gram-positive action, containing penicillin-sensitivity and penicillin-resistance S. pneumoniae. The second-generation of piperazine groups of the fluoroquinolone drugs improved their Gram-negative activity, whereas their alkylated counterparts improved their Gram-positive activity. In this site, an alkylated piperazine group functioned similarly to a pyrrolodinyl group. It has been shown that altering the R5 position enhances activity against Gram-positive bacteria. The following sequence was demonstrated to improve antibacterial efficacy: [13] -CH3, -OH, and -NH2. Sites R (eight), R (five), and R (seven) pointed at the 3rd generation were modified to improve activation against Gram-positive bacteria. The R7 tuning has shown to be the most effective of these modifications. This may be shown by contrasting the MIC90 of those medicines. With a dimethyl pyrrolodinyl group at R (seven) and Cl2 at R (eight), clinafloxacin is considered to have the third-generation medications with the highest promise. In this group, clinafloxacin had the lowest MIC90 (Table 3). Despite having similar structures, ciprofloxacin and sparfloxacin are more potent when combined because of the inclusion of amines at R (five) and the alkylation of the piperazineset (Table 3). With the exception of the replacement of the -CH3 in the case of grepafloxacin, it is identical. All of the third-generation requirements and action against anaerobic organisms are included in the range of activity of the fourth generation chemicals. The improved activity against anaerobes is caused by N2 atom at the R (8) location, and the addition of a 2,4-difluorophenyl set at the N place increases the total efficacy of the drug. This shift is visible in the moxifloxacin, gemifloxacin, and trovafloxacin structures (Table 2). The pyrrolidine at the R (seven) location has been changed, and an azabicyclicset and the large partseries have also been added both of which increase gram-positive activity [14] include the totaling of difluoromethyl ether set at the R8 point. When moxifloxacin and gatifloxacin's effectiveness and structures were evaluated, the azabicyclic at the R (seven) position formed the most potent effect against Gram-positive bacteria. The R7 location is the only structural difference between these two molecules. The azabicyclic grouping in moxifloxacin greatly increases gram (plus) potency in comparison to gatifloxacin (Table 3).
Complexation process of quinolone according to chemical properties
Because they include a basically piperazinyl rings (or an additional N-heterocycle) at place seven and a carboxylic acid action location on site three, quinolone molecules are often zwitterionic. Both effects are minor, and the quinolones are well soluble in either acidic or basic conditions. Potentiometric, 1H-NMR spectroscopy, and the UV spectrophotometer have all been used to explore quinolone protonation equilibrium in an aqueous solution [15]. The general structure of a quinolone molecule given in may allow the discovery of two proton-binding sites (Scheme 4). Such a molecule possesses two protonation isomers among its four microscopic protonation forms in solution.
Table 3. Proportional MIC90s of quinolones. The effectiveness of the medicines existing in MIC90 (mg/L) of every treatment on changed Gram-negative straining and Gram-positive strains
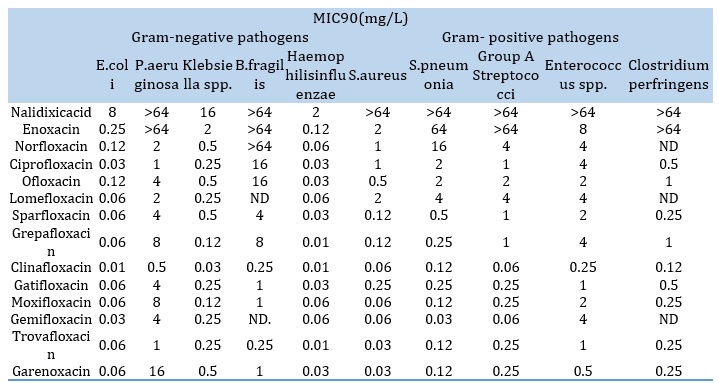
The molecular and sub-molecular properties of acid-base are depicted by using themicrospeciation of pharmaceutical compounds (micro constants). The total basic behavior of the molecules is measured by the macro constants. However, the readings for pKa2, which correlates with the fundamental function of piperazinic group, beginning from 7.57 to 9.33, those for pKa1, which corresponds to the acidity of the carboxylic group, range from 5.33 to 6.53. Table 4 displays the protonation the constant values for two representative quinolones, ofloxacin and norfloxacin.
The micro constants, which represent the separate protons-binding affinities of the functional groups and depend on pH, are used to compute the concentrations of various protonation isomers. Between pH 3 and 11, quinolones mainly take the zwitter ionic form. 99.9% of the positive charge formula QH2+ which is existing at pH 1. At pH 7.4, the abundance of every micro species is comparable. Quinolone bioavailability has been connected to quinolone microspeciation, antibacterial activity serum protein binding, and compounds. Molecules, which act as ligands in the Themicrospeciation of quinolone non-dissociated shape of (Q) in basically environments also in azwitter ionic formula (QH) in the neutrality, a little acidic, and basic conditions media that is a little simple. In very acidic environments, quinolones can be converted into their cation form, QH2+, to produce ionic compounds. Because they may be binding to the metallic ions, quinolones produce metal complexes. Quinolones can function in metal complexes as a bridging, unidentate, or bidentate ligand. One of the deprotonated -COOH collection O2 atoms and the ring -C=O oxygen are widely used in quinolones to form bidentate coordination atom (Scheme 5a). When coordinated from side both piperazinic and N2 atoms, quinolones can function as a bidentate ligand (Scheme 5c). When connected to metal ions by terminal piperazinyl nitrogen, quinolones have ability of complexes formation as unidentate ligands (Scheme 5d). In a solid state, polymeric complexes have various coordination types accessible. When conditions are extremely acidic, quinolones protonate, and show up as cations in ionic complexes.
Metal complexes of quinolones
Metal-quinolone chelates
Two major metal chelate-forming sites are presented in quinolone molecules (Scheme 6). The most common kind of coordination in quinolone chelates is the first one symbolized by close-by carbonyl and carboxyl groups. Mg2+, Ca2+, Cu2+, Zn2+, Fe2+, Co2+, and other divalent cations can be bound by quinolones. Creating chelates with a metal: ligand stoichiometry in apportion of 1:1 or 1:2, or cations having valence of three (A13+ and Fe3+), generating bounded with a metal: ligand stoichiometry of 1:1, 1:2, or 1:3. (Metal: ligand stoichiometry). Complexes containing Bi3+ have a greater stoichiometry (1:4). The overall structure of quinolone chelates with cations which consider as divalent with a molar ratio of 1:2 (ligand: metal) is presented in (Scheme 5). The number of coordinated ligands was shown to be pH dependent on a learning of the copper-ciprofloxacin organization. Thus, a 1:1 complex is preferred in the more acidic zone, while a one: two complexes is the chief types at greater limited pH [16].

Scheme 4. Protonation scheme of a fluoro-quinolone molecule with piperazine ring at the 7-positio
Table 4. Values of the norfloxacin and ofloxacin protonation constants

Quinolones were discovered to have a comparable trivalent cation (Al3+ and Fe3+), which have a strong connection for metal ions, can form highly stable chelates by using strong Lewis acids. Chelates containing group 2A cations (Mg2+, Ca2+, and Ba2+) are less stable. For ciprofloxacin chelates, for example, the formation constant values drop in the following direction: "Al3+> Fe3+> Cu2+> Zn2+> Mn2+> Mg2+". The variance for norfloxacin chelating is attractive similar to ferric>aluminum>copper>frrous>zinc>magnesium>calcium. The strength of the chelate's stability depends on the solvent's pH and as dependent on the dielectric constant; the lomefloxacin's affinity in these ions Ca2+ and Mg2+ reductions in the following: Cation> anion >zwitterion> [17]. Tables 4, 5, 6, and 7 display several chelates created in a solid shape by using quinolones as bidentate ligands with one carboxylate oxygen, one pyridone oxygen, and kinds of performed experiments to look into their biological activity. The quinolones are considered to be bidentate ligands in the chelates; complexes having another bidentate co-ligands (e.g., 2,2'-bipyridine, 1,10-phenantroline) which have biologically in action never explored here.

Scheme 5. Chief management types of quinolones

Scheme 6. The overall building of 1:2 (metal: ligand) quinolone chelating by using cationic bivalency
Table 5. Selection of chelate quinolones as a chief formation

Table 6. Certain quinolones chelation in the second formation

Table 7. Choosing the chelating of quinolones at three and four generations

Chelates presentedin the surface of polyoxometalates (POMs)
To create metal-organic polymers with therapeutic applications, quinolone molecules make suitable multidentate ligands because of the increasing density of electronic clouds of O2 and N2 atoms [18]. Such hybrid organic-inorganic molecules were produced by chelating quinolones onto the external of polyoxometalate (POMs) anion recognized as anti-tumors, antivirals, and antibacterial inorganic medicinal compounds; the goal of external modification with biologically active chemicals is to enhance their effectiveness. A quinolone hydrothermally reacts together with metallic salt and a polyoxometalate in the heat presence was the main method used to produce these compounds (in acidic or ammonium salt form) when changing pH of V4O10(2-O)2[VO(H-Cf)2)]. One of the most basic substances in this family is 2•13H2O, which has a structure made up of single V4O12 1 unit and 2 corner-dividing octahedral VO6-ciprofloxacin parts connected by two 2-O channels [19]. Compounds consisting of PW12 or SiW12 groups and double M(Quin)2 chelates were constructed using anions with a -Keggin structure (PW12O40, SiW12O40). The quinolone molecules and PW12 or SiW12 clusters behave as an organic ligand that chelates bidentates to support the metal ions (Scheme 7). Scheme 7 depicts a 1D chain architecture made up of POM clusters and binuclear metal clusters coupled to the unidentified or bridging bi-dentate inorganic ligands, POM clusters. The coordination of the metal ions is carried out by the quinolone molecules with PW12 or SiW12 clusters acting as chemical ligands with chelating properties (Scheme 7).
Quinolone operating as a unidentate ligand in metal complexes
The terminal piperazinyl nitrogen (N4) of quinolones with a piperazinylcircleat the 7-place may participate in the coordination of metal ions. When the transitional metals like silver, gold, and ruthenium are combined, this coordination state has been seen (III). The substance Ag2(Nf)2(NO3)2's putative structure is depicted in (Scheme 8) [20].

Scheme 7. A binuclear metallic cluster of quinolones bound to POM clusters
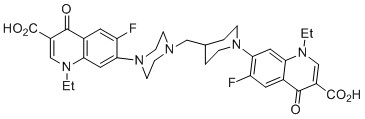
Scheme 8. Suggested forming of the Ag(H-Nf)2(NO3) complex

Scheme 9. Expected shape related to a) Ag2(Nf)2(NO3)2 and b) [Au(Nf)2(H2O)2]Cl3
Scheme 9a,b reveals the mononuclear complex [Au(Nf)2(H2O)2]Cl3 and the di-nuclear complex Ag2(Nf)2(NO3)2 that were produced because the interaction of silver and gold with norfloxacin [21].
Polymeric complexes
To produce the dimer complexes [Mg2(H2O)6(HNf)2]Cl4.4H2O and [Ca2(Cl)(HNf)6]Cl3.10H2O [22], norfloxacin functions as a bivalent bridge ligand linked by the oxygen from pyridone and one from a carboxylate (unidentate bridging) (Scheme 10). The substance [Pb(H- Nf)(ONO2)2] was found to have similar coordination (Scheme 11).
Cd2(Cx)4(H2O)2: di-nuclear complexes X-ray crystal structure determination [Cd2(Cx)4(DMSO)2] and 10H2O, 2H2O. The Cd+2 is coordinated with seven water molecule presented in the coordination environment, 2 cinoxacinate ions serve as bridge binders with tridentate chelates, and two cinoxacinate ions performing as bidentate chelate ligands. In polymeric complexes, many kinds of coordination may simultaneously exist. Norfloxacin adopts many kinds of coordination when two Fe(II) Depending on how the synthesis was conducted, complexes were presented. Scheme 12a demonstrates the structure of Fe(II) in the presence of two norfloxacinate anions connected together in a bidentate ligands arranged during the pyridone oxygen, one carboxyl carboxylate oxygen, and two molecules of norfloxacin bound as unidentate ligands organized by double O2 atoms since multiple opposite carboxylates. Scheme 12b [23] demonstrates that in another complex, Fe (Nf) 24H2O, there are double molecules which are bonding as bidentate ligands and possibly double as unidentate ligands.

Scheme 10. Structural form of the Dimeric complexes [Mg2(H2O)6(HNf)2]Cl4⋅4H2O

Scheme 11. Building shape of the dimeric complex [Pb(H-Nf)(ONO2)2]2

Scheme 12. Coordination modes of norfloxacin in a) Fe (H-Nf)2(SO4). 2H2O and b) Fe(Nf)2⋅4H2O
The pseudo-tetranuclear basic elements of a 1D ladder-similar Ag(I) organization polymer [Ag4(H-Cf)2(Cf)2(NO3)2].4H2O [24] are generated by the combination of tetradentate deprotonated ciprofloxacin ligands and unidentate ciprofloxacin are linked by the N4 piperazine atom (Scheme 13).
Ionic complexes
According to the essential activity for the N4pyrazinyl, in an acidic environment, quinolones undergo protonation in the environment to generate ionic chlorometalates that are usually created by the gradual evaporating of a salt solution complicated with metal in nature. This complexation was primarily evaluated for their antibacterial efficacy (see subsection 4.3). The chloroantimonates (III) produced from the ciprofloxacinium ions and nalidixiumcation (C12H13N2) typically have the following format: (C12H13N2O3)[SbCl4]H2O, the ciprofloxacin cations (Cfh3)2+), and [SbCl5]H2O. Two distinct forms of chlorobismutates (III) were produced thanks to ciprofloxacin: (CfH2)(CfH)[BiCl6]2H2O and (CfH2)[Bi2Cl10].4H2O [25]. The tetrachlorocuprates (II) produced from norfloxacin, pefloxacin, and cinoxacin are designated as (NfH2)(NfH)[CuCl4].ClH2O[CxH2] [CuCl4]. The analogous chemicals are H2O and (C17H22FN3O3)2+[CuCl4]-2.
Enrofloxaciniumtetrachloroferate(III) and (erxH2)[FeCl4]Cl are two more chloromethalates [26], ciprofloxaciniumtetrachlorozincate (II) dihydrate [C17H19N3O3F], and the latter compound. Ciprofloxacin tetrahydrotetrachloroaurate, 2[ZnCl4] 2H2O(III), (cfH2)[AuCl4]H2O, and the trihydrate of ciprofloxaciniumhexachlororuthenate (III), (cfH2+)3[RuCl6]3H2O were also observed.

Scheme 13. Direction type of ciprofloxacin's anion in {[Ag4(H-Cf)2(Cf)2(NO3)2]⋅4H2O}n
Mechanism of action of quinolones
In the relation of facility of quinolone complexes that attach to DNA was investigated to better understand how quinolones work. In contrast to free quinolones, the quinolone-Mg2+ combination interacted with DNA and gyrase, according to the experimental results, a ternary configuration has also put out. Because of that, the C=O and COOH moieties of norfloxacin serve as a link among the phosphate class of the nucleic acid and these molecules, and further stability is the condensed rings of antibiotic and also the base sequences of DNA combine with one another in stacks to provide. To construct a the triple Cf-Mg2+-duplex adduct is modeled. Direction the communication of an oligonucleotide double with ciprofloxacin was examined in both the loss and presence of Mg2+. The arrangement of CFX and Mg2+ in the minor groove of DNA was conserved by docking on this model. The following quinolone-divalent metal I associations were used to evaluate the in vitro interactions with calf thymus DNA: norfloxacin-Cu2+ [27], ciprofloxacin-Mg2+, -Cu2+, levofloxacin-copper(+2),"gatifloxacin-magnesium(+2), copper(+2), cobalt(+2), cadmium(+2)" [28], and flee. In accordance with the experimental evidence, the metal ion mediates the connection involving quinolone, DNA, and the ability of the quinolones of metal complexes to engage in active site bonding with DNA. Tests performed in vitro revealed that the compressed rings of antibiotic and the base sequences of DNA react with one another in stacks to provide. Nevertheless, in the Mg2fle presence, the quinolone-gyrase-DNA complex develops DNA can be contacted by the metal complex of quinolones through an intercalative binding process. According to experimental evidence, and the metaling ions act as a medium in the interactions in both quinolone and DNA [29]. Tests conducted in vitro revealed that, while DNA gyrase may connect to quinolones in the presence of DNA. Furthermore, when Mg2+ is presented, the quinolone-gyrase-DNA complex develops. Four water molecules and two C3/C4 O2 atoms from a chelated quinolone make up the proposed Mg2+ attached to topoisomerase IV's coordinating environment. On the side chain of serine, the hydroxyl and carboxyl group that in side chain of serine glutamic acid are connected by hydrogen bonds formed by two of these water molecules. It was suggested that this water-metal ion "bridge" might aid the quinolones and topoisomerases contact between them [30]. Mutations in one or both amino acid residues, which partially or totally disrupt the bridge function, and subsequently the interaction between the protein and the quinolone, are the main factors that contribute to quinolone resistant.
Bioactivities of quinolones
Unquestionably, quinolone-anchored natural products and synthesized compounds have demonstrated a wide-ranging in biological or pharmacological action. Antibacterial, antioxidant, anticancer, anti-inflammatory, anti-malarial, anti-fungal, and anticancer actions are only a few of them [31].
Antibacterial activity
Desai et al. created the quinoline compounds with the highest antibacterial potency, numbers 1, 2, and 3 (Scheme 14). The substances' ability to fight off Pseudomonas aeruginosa, Escherichia coli, Streptococcus pyogenes, and Staphylococcus aureus was assessed in the present of ampicillin as a reference. The results show this substance has powerful antibacterial action even at the modest inhibitor doses of 12.5 mg/mL and 50 mg/mL. The potential activity of these compounds, according to the authors, is directly correlated with the substituent effect on the ring. Le et al. claim that hydrazone is used to link these three bioactive quinoline compounds [32] to hydrazone-containing derivatives of quinolone. Fu et al. also created the Quinoline byproduct 7 hybridized with a piperazine moiety linkage and noticed wide-ranging-spectrum antibacterial action on diverse the MIC levels of microorganisms ranging from 0.125 to 8 mg/mL [33]. The development of the targeted bacteria was effectively prevented by linkers 4, 5, and 6.
Antioxidant function
Comparing derivative of quinoline 114 coupled with a -aminophosphate to regular DPPH, Bazine et al. found that it displayed substantial antioxidant action (Scheme 15) [34]. By adding a phenol circle as an alternative to the quinolone scaffolding, the scientists showed that the bioactivity was further changed.

Scheme 14. Chemical compositions of quinolone derivatives with antibacterial activity
Anticancer activity
The anticancer efficiency of the quinolone compound 8 against neuroblastoma cells was established by Bingul et al. and confirmed (Scheme 16). The researchers found that compound 8 significantly reduced the viability of neurocancer cells, while also showing strong anticancer efficacy against the Kelly neuroblastoma cell lines and SH-SY5Y [35]. Compounds 10 and 11, according to Othman et al. are quinoline derivatives with a thiophene molecule anchoring them. With corresponding IC50 values of 38.41 and 28.36 M, the quinolines previously described showed a strong antitumor effect against the human cancer cell line MCF-7. In addition, Kundu et al. found that the most effective treatment for human topoisomerase 1 was quinoline 9 in combination with an imidazole and 1,3,4-oxadiazole [36].

Scheme 15. Chemically form of an antioxidant activity of quinolone derivatives

Scheme 16. Chemical compositions of quinolone extracts with anticancer activity
Activity agents Anti-inflammatory
Indeno [1, 2-c] quinoline products 12 were developed by Tseng et al. who characterized them as strong anti-inflammatory drugs with low cytotoxicity that are also efficient against tuberculosis (Scheme 17) [37].
Antileishmanial activity
Upadhyay et al. created a quinoline derivative 13 that was triazole-anchored and discovered with an antileishmanial action (Scheme 18) [38]. The researchers found that the activity of the synthesized molecule was enhanced by the addition of a chloro-substituent. A hybrid antileishmanial agent with remarkable efficacy is phosphorylated quinoline 14. The basis for a potent antileishmanial activity may lie in phosphorus-quinoline hybridization.
Antimalarial activity
Researchers are now looking for ways to enhance and boost the antimalarial activity of medications made by using quinoline scaffolds. Most of them are synthesized quinoline derivatives that have been combined with readily available, potentially identifiable medications [39]. The researchers hypothesize that hybridization will save money and lessen the risk of drug side effects. Quinoline-artemisinin drug hybridization was described by Lombard et al. who also providingcomposite 15 (Scheme 19). The hybrid compound has an excellent antiplasmodial action, while not having as much antimalarial activity as dihydroartemisinin. The hybrid quinoline-sulfonamide derivative 16, which has antimalarial properties, was produced by Verma et al. [40]. The hemozoin formation was prevented by the hybrid chemical, as the authors found.
Antifungal activity
6-Perfluoropropanyl quinolines 17 and 18 were created and described by Fang et al. to be antifungal active substances. (Scheme 20). The antifungal activity of the generated quinoline compounds against Pyricularia oryzae was astounding. A pyrazole-quinoline hybrid that is antifungally effective was developed and reported by El Shehry et al. (Scheme 20) [41]. The substance was created and indiciated significant antifungal effectiveness against by the intended fungus kind.

Scheme 17. Structuring of anti-inflammatory for activation quinolone driveling
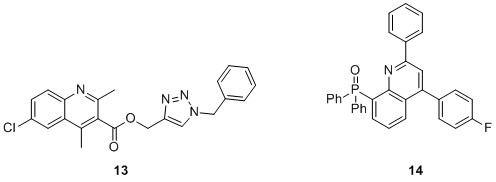
Scheme 18. Chemical structure of antileishmanial active quinoline derivative

Scheme 19. Chemically structuring of hybridization of quinolones

Scheme 20. Chemical compositions of quinolone derivatives with antimicrobial effects
Conclusion
Antibiotics called quinolones consider as a class of synthetic bactericidal has a wide range of activity and has the ability to inhibit both Gram-(+) and (-) bacteria together as well as anaerobes. They affect the DNA synthesis process by adhering to bacterial topoisomerase form II enzymes. The quinolone's class of ketones carbonyl indirectly binds to the enzymes' serine in the acid residues through the Mg2+ ion during bonding to the breakdown complex. The R1, R6, R7, and R8 positions of quinolones can be altered to enhance action, pharmacokinetics, and hazard. The ideal substituents include cyclopropyl groups at R(one), fluorides groups at R(six), azabicyclic groups at R(seven), and OCH3groups at R(eight), despites this, the F2 being much more abundant at site R6. The amount of innovative analogs in the clinical pathway demonstrates this. It is clear that anti-resistance and interactions advancements are definitely possible, and new quinolone generations can continue to contribute to the effective management of bacterial illnesses. Quinolone and its compounds have demonstrated promise in the behavior of a range of human diseases, like cancer, malaria, fungal infections, and bacterial infections. The aforementioned green synthesis techniques are frequently advised for the creation of this magnificent organic compound and its variants.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Orcid
Aseel H. Abad Al-Ameer 0000-0001-9291-3568
How to cite this manuscript: Aseel H. Abad Al-Ameer*. Quinolone antibiotics and their applications in metal complexes: An update. Asian Journal of Green Chemistry, x(x) 2022, xx-xx. DOI: 10.22034/ajgc.2022.3.4
)