Document Type : Review Article
Authors
1 Biology Department, College of Science, Al-Muthanna University, AL-Muthanna province, Iraq
2 Chemistry Department, College of Science, Al-Muthanna University, AL-Muthanna province, Iraq
Abstract
This review is mainly focused on the recent development of rice husk utilization for the efficient immobilization of organic molecules. It was pointed out that sodium silicate precursor was prepared in large amounts from rice husk ash. Moreover, a new route was designed for the extracted sodium silicate through a purely homogenous method in-situ reaction of 3-(chloropropyl)triethoxysilane (CPTES) with silica. The product was solid silica with chloride end groups. The silica-chloride end groups are the cornerstone for the design of heterogeneous catalyses with the specific functional groups for different purposes. These functionalized silica catalysts have attracted widespread attention due to their good physical and catalytic properties in a large variety of chemical reactions.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
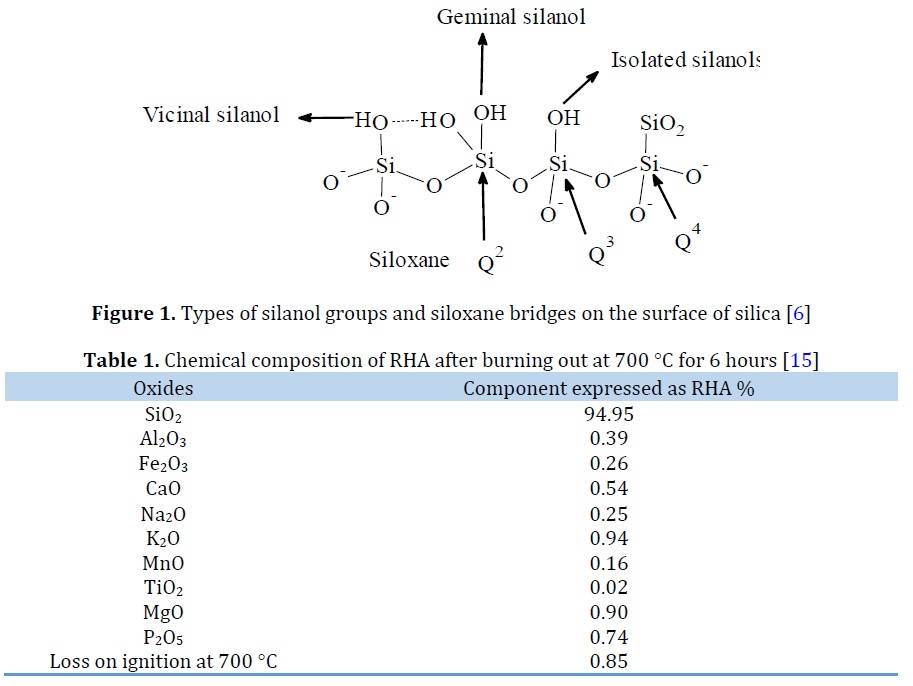
Zhuravlev [1] and Vansant [2] reported that research on silica began in the1930 s when many scholars investigated the condensation processes of silicic acids. These studies led to the discovery of the surface silanol groups, ≡Si–OH. Infrared spectroscopy analysis which was used by Yaroslavsky showed the major adsorption bands due to the existence of hydroxyl groups on the silica glass surface [1]. Due to various spectral and chemical analyses, it has been known that silica contains two types of functional groups, i.e. the siloxane (≡Si–O–Si≡) inside the matrix of silica and OH groups (silanol groups) on its surface.
There are two free silanol groups present on the surface: the isolated silanol (≡SiOH) (a silicon atom bonded to a single hydroxyl group, and the geminal silanol (=Si(OH)2) (silicon atom bonded to two hydroxyls group, as displayed in Figure 1. It was found that each two vicinal silanol groups can form a hydrogen bond [4]. Likewise, silica contains three kinds of siloxane groups (≡Si–O–Si≡). These siloxane groups (≡Si–O–Si≡) are represented as Qn according to NMR, where "n" is equal to bridging bonds (–O–Si) bond to the central Si atom, i.e. Q4–silicon atom bonded to other silicon atoms via four bridging, while Q3– represent three siloxane groups inked to the single silicon atom and Q2 – two siloxane bonds connected by one silicon atom as depicted in Figure 1. Generally, siloxane groups number can be calculated from the following equation: Qn = Si (OSi)n(OH)4-n [5, 6].
Rice husk (RH) is a biomass material which recycled successfully [7] More than 75 countries are produced rice [8]. Production volume of rice worldwide reached 400-545 million metric tons annually, where the husk is about more than 20% of the total production [9, 10]. The husk content is high in silica, and therefore it has economically valuable. The low cost of husk makes it an attractive raw material for utilizing [11]. About 20% of silica powder can be extracted from RH and widely used for various applications instead of the commercial silica product [12].
The cellulose is about 38% in rice husk followed by lignin at 22%, while silica is about 20% and 18% of pentose. The other 2% is inorganic elements. Rice husk as in the other agricultural waste, when it was burned could cause environmental pollution. Therefore, the control of temperature and pressure are important factors of the minimizingollution [11]. The ash of rice husk (RHA) is the main product of the calcination contains 95% of pure silica [13]. An analysis was made for the RHA components which were burnt at 700 °C, as reported in Table 1 [14].
Experimental
Silica extracted from rice husk
After collecting RH, it was cleaned with water a few times to remove dust and then dried at laboratory temperature for 24 hours. To remove metal oxide, the RH was treated with one molar nitric acid in a plastic container for 24 hours, and then washed more than six times with distilled water. Afterward, the leached RH was subsequently oven-dried at 100 °C for 24 hours, followed by combustion at 800 °C for 6 hours. The high-purity white rice husk (RHA) was obtained after washing with distilled water and drying. A fine powder obtained is ground using a mortar and pastel to be used as a silica source [14, 15].
RHA Functionalization with CPTES
The silica from RHA was dissolved in one molar sodium hydroxide and then filtered to isolate undissolved particles. A certain amount of CPTES was added to the sodium silicate solution. The mixture was stirred continuously and gradually titrated with 3.0 M of nitric acid in the presence of a pH meter. A white gel began to form when the pH values lowered to slighter than 11.0. The titration was resumed until the pH of the solution decreased to 3.0. The gel aged for two days at laboratory temperature. The fresh gel was separated by centrifugation and purified via repeated washing with distilled water. The last washing was performed by acetone as a volatile solvent to enhance the drying product. Finally, grind to produce white powder labeled as RHACCl.
Immobilization of RHACCl with organic molecules
The synthesized solid RHACCl has been utilized as a support to immobilize functional organic molecules onto silica surface. The modification was organized mostly by grafting organic ligands on the silica framework by replacing chloride end groups in RHACCl. The mixture was refluxed at 110 °C in an oil bath for the necessary time. Then, the composite was isolated and washed with the fresh organic solvent. The dried sample is necessary dried at 100 °C. The obtained sample was labeled relying on the organic ligand utilized.
Results and Discussion
New route for the production of silica
Two processes of producing silica are discussed in this review. In the first assay, rice husk was acidified with nitric acid, filtered, and then burnt in a furnace at 800 °C [14], as shown in Scheme 1a. This fast process produced pure silica from this biomaterial waste effectively which facilitated using organic ligands for various applications [15, 16]. On the other hand, one of the disadvantages of the methods was environmental pollution caused by releasing many toxic gases and acidic oxides. In the second procedure, the RH was treated or washed with nitric acid, and then the RH was neutralized using NaOH, as displayed in Scheme 1b. After that, the silica was formed by titrating sodium silicate solution vis HNO3. The main target of this procedure was producing silica capable for incorporating with metals such as cobalt, vanadium, iron, and so forth [17, 18]. The limitation of this method was that the silica produced may have organic impurities due to use NaOH. It has been established that these organic impurities will interact selectively in the material characterization, especially if the silica has been conformed to the specific organic ligands.
It had been observed that many organic acids and esters easily reacted with sodium silicate in a one-pot synthesis. These compounds are acetic acid (AA), chloroacetic acid (CA), chloroethylacetate (CEA), 1-chlorohexane, and ethyltetadecanoate (EP). The morphology study shows that the nanoparticles have sphere shapes [19] The chemical synthesis reactions are depicted in Scheme 2. The results demonstrated that the produced biomolecule silica was successfully modified by acid or the ester functional ends groups and not reacted with the halide functional end groups. It was also disclosed that the silica could not assemble with an alkyl halide (1-chlorohexane) due to the lack of carboxyl or ester functional end groups. The effective procedure used in this study was environmentally preferred, easy, and yielded silica connected to chloride or alkyl end groups.
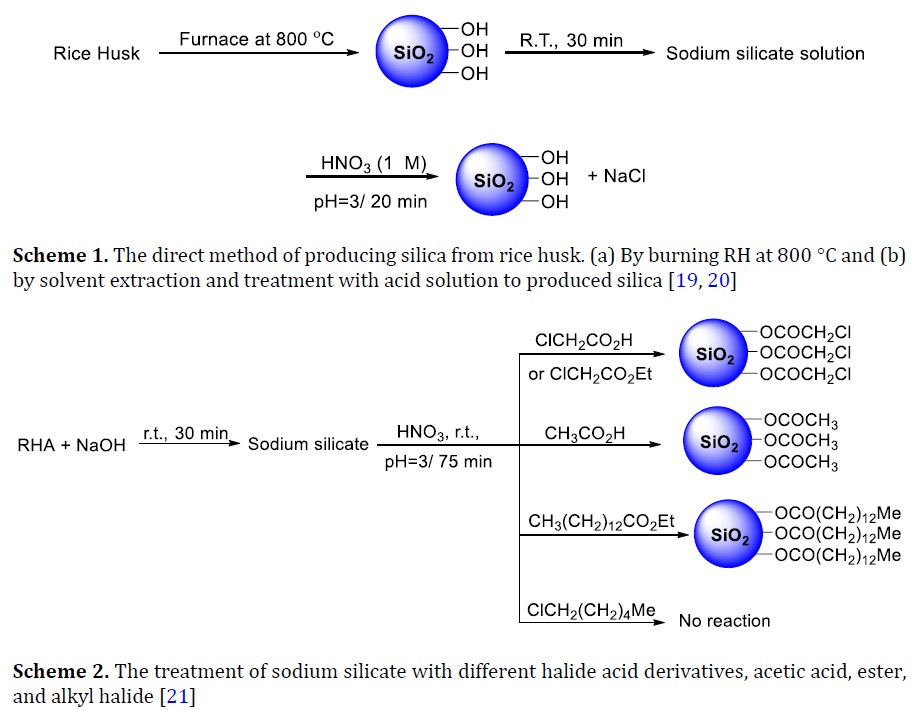
Figure 2a and b [21] shows the N2 adsorption-desorption isotherms and pore size distribution diagrams for silica-chloroacetic acid (RHACA), and silica-chloroethylacetae (RHACEA). All the isotherms were type IV and exhibited H2 hysteresis loop [20] The very high surface area was deducted for both RHACA and RHACEA which was 655 and 654 m2gm-1, respectively. RHA had only 347 m2gm-1 specific surface area. Thus, the new method of immobilizing silica with those ligands could reach the products via a pure homogeneous procedure which may enhance the specific surface area and mesopores structure of RHACA and RHACEA. Adam et al. [21] reached a similar conclusion when other organic ligands were incorporated inside the silica matrix. The pore size distribution was found in a range of 4 to 8 nm with pore volume 4.5-4.2 cm3gm-1 suggesting mesoporous silica structure (Figure 2a and b). Both chloroacetic acid and chloroethylacetate had BET the same surface area. The corresponding N2 adsorption-desorption and pore size distribution curves indicated the formation of the mesoporous system (inset in Figure 2c and d) for RHAA and RHAEP, respectively. The isotherms were type IV and exhibited the H2 hysteresis loop. About 539 and 399 m2gm-1 were deducted as a specific surface area of RHAA and RHAEP, respectively, while pore diameter for RHAA and RHAEP (Figure 2c and d) showed an in the range of 4 to 8 nm. About 7.2-3.9 cm3gm-1 was the average pore volume for RHAA and RHAEP which was fall in the mesoporous range. The lower specific surface area was deducted for RHAEP as a result of the immobilized large molecule into the silica. RHACH had adsorption isotherms of nitrogen similarly sigmoidal to RHA.
Very regular and spherical-shaped particles were found in TEM micrograph for both RHAA and RHAEA, as indicated in Figure 3 [22]. The measurement of particles by TEM found that the particles had 5 nm in diameter.
In our laboratory, it was recently reported as a simple method to modify silica [22] similarly to the method in Scheme 1b with a slight modification. After generating sodium silicate, it was burnt at high temperatures to separate the co-deposited carbon residue during frequent extraction. This will mimic and avoid the atmospheric pollution caused by organic released when silica is burnt in the furnace. The prepared silica will guide to design useful heterogeneous catalysts conjugated with metals or with organic ligand grafting in various applications. In other cases, the silica produced can be desirable in the chromatography column technique to separate, and purify constituent particles of a mixture of organic substances [19].
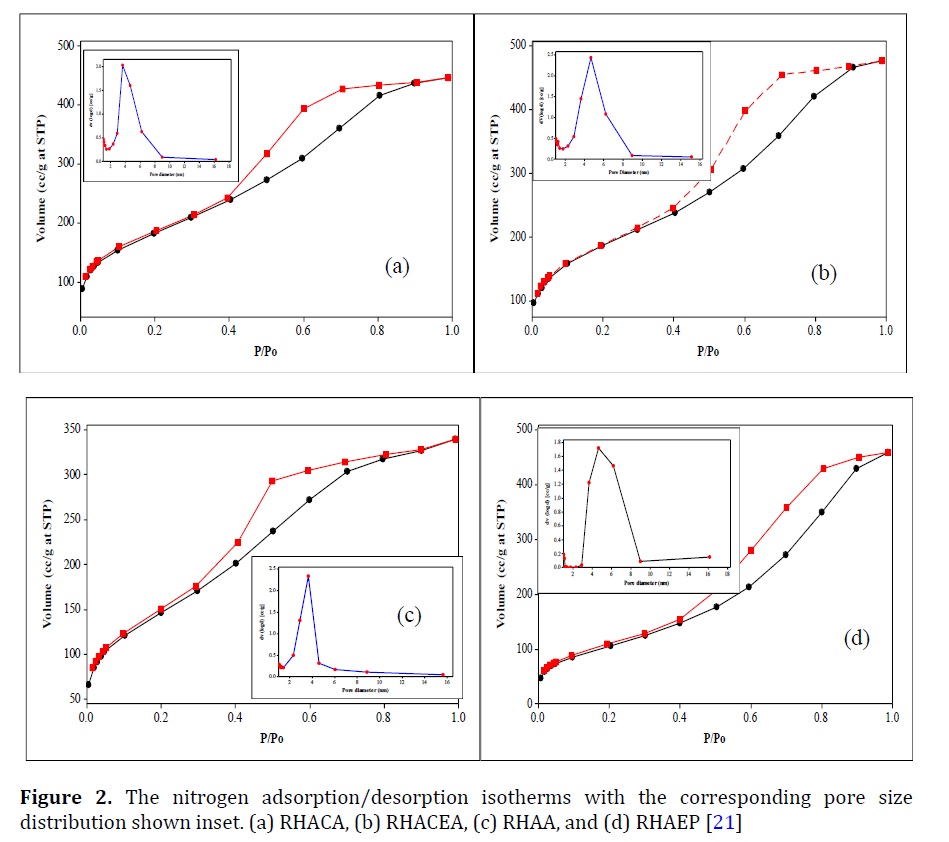
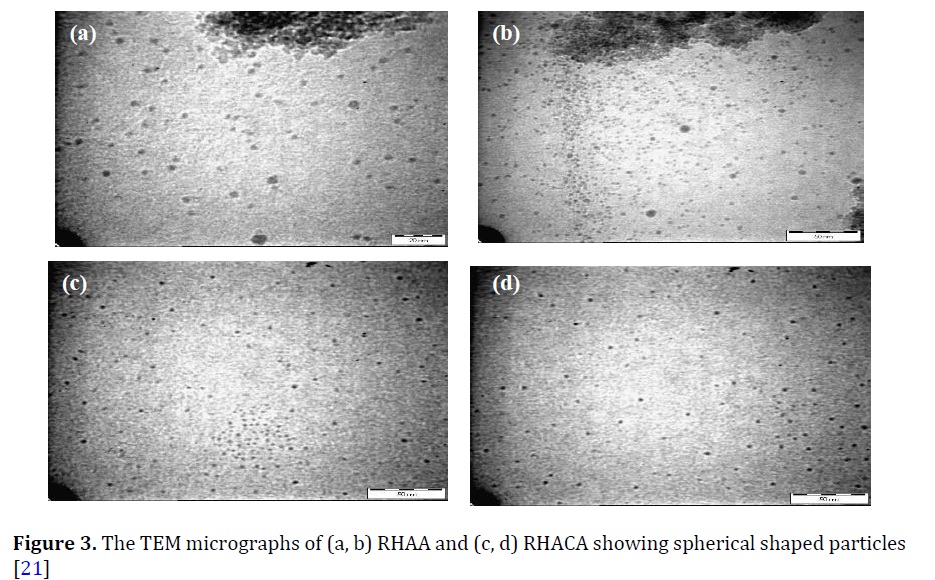
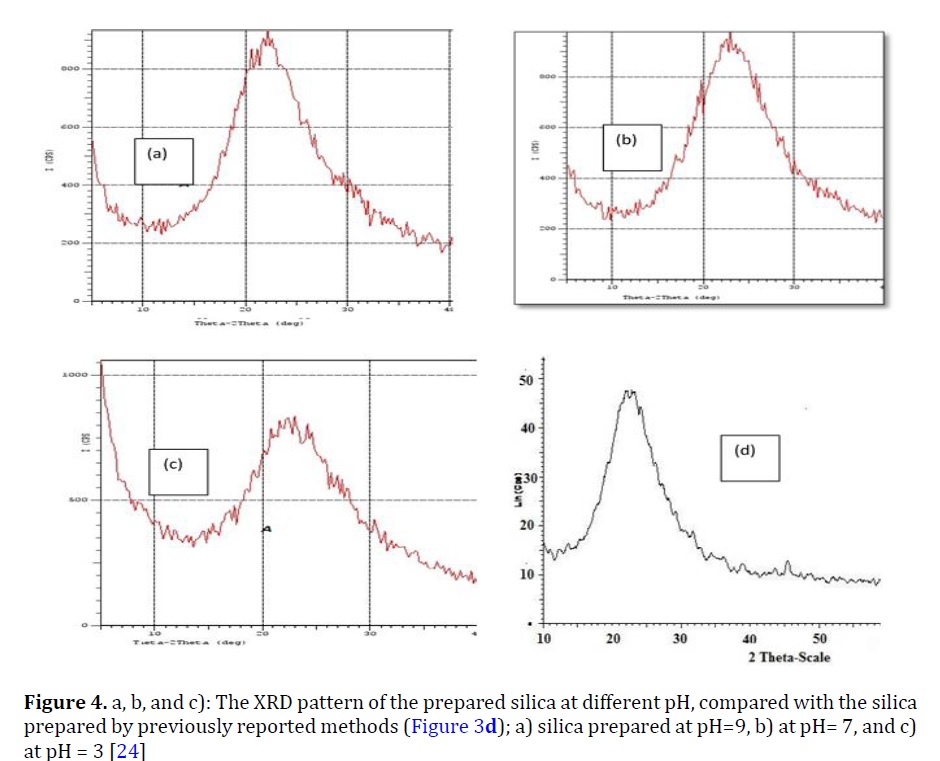
Figure 4a, b, and c show the X-ray powder diffraction (XRD) pattern of the modified silica by the procedure designed [24] compared with the silica prepared by the other techniques documented in the literature (Figure 4d). All the diffraction patterns were typical of the amorphous silica.
Organic-inorganic hybrid catalysts
Recently, a great deal of attention has been devoted to the modification of the silica surface [23-24]. This process can be directed by many researchers toward managing and adjusting the chemical effects and technical characteristics of the hybrid material. Different catalysis can be prepared by modifying the silica surface. These could be the promising nanostructured silica materials, selective heterogeneous catalysts, and liquid crystals phases [25].
The functionalization of silica surfaces is mainly employed using organic ligand molecules. Silica-silylating hypered have been found mostly chemically reactive [26]. (RO)3Si–R* is the general formula of silylating agents. The R is represented methyl or ethyl groups, while R* is n–propyl with the terminal functional group [27].
In this method, a mixture of tetraalkoxysilane Si(OR)4 and the silane coupling agent (RO)3Si(CH2)3X are combined in an alcoholic solution in both acid and base catalyst, where simultaneously hydrolysis and condensation reactions proceed [28].
A silylating agent immobilized onto a silica surface has been considered as a good entry to introduce basic groups allowing anchored pendant chains [29]. Using a silica-silylating hypered catalyst, it was found that the catalyst prevents the removal of the organic functional group from the surface by different aqueous or non-aqueous solvents [30]. It also controls thermal and hydrolytic stability [31, 32].
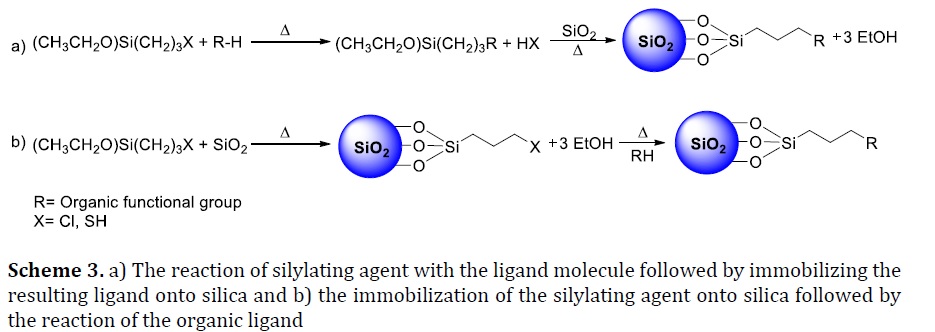
Silica-silylating hypered were prepared by two procedures. The first strategy is to branch covalently silylating agents and the ligand complex, supported by pre-extracted silica in a heterogeneous reaction, as depicted in Scheme 3a. The second process is to add the synthesized polysiloxane to the complex group, as demonstrated in Scheme 3b. Both these processes have been widely researched [33-37]. It was noticed that these techniques contain longer preparation times, and commonly used hazardous chemicals often resulted in inefficient preparation methods. Therefore, there was a need to design a simple and more direct method to prepare the immobilized silica with silylating agents.
CPTES immobilized onto silica was improved by solid-liquid mixed phase reaction (heterogeneous reaction). Bae et al. [27], Hoegaerts et al. [39], Shi and Wei [40] showed that the treatment requires reflux in toluene for 24 hours. This step was followed by soxhlet extraction using various organic solvents such as toluene and dichloromethane. It was also found that the pre–dried silica at 150 °C for 12 hours under 10 Torr led to functionalized silica CPTES in toluene under reflux conditions for 14 hours [26]. 3–(chloropropyl)trimethoxysilane can also functionalize surface of silica at 150 °C under reflux conditions for 72 hours, as mentioned by Alcântara et al. [33].
Recently, a simple procedure was presented by Adam et al. [23] to prepare silica-silylating hypered. The procedure followed pure homogenous rote in a one-pot synthesis, as indicated in Scheme 4. The silica-silylating hypered was labelled as RHACCl.
The FT-IR spectra of silica (RHA) with RHACCl compared with the differential spectrum were illustrated in Figure 5 [24]. In FT-IR, Si–O–Si bands were located at 1101 cm−1 [34]. In RHACCl spectra Si–O–Si bands were shifted to 1069 cm−1. The absorption at 1040 cm−1 in the differential spectrum could be for the –Si–O–Si– and –Si–O–Si–C.
At 3471 cm−1 the absorption peak is usually to O–H vibrations. Both the silanol group and physically adsorbed water may give this peak [35]. The absorption at 2959 cm−1 could be for the C–H stretching vibration. The CH2–Cl bond appeared at 698 cm−1. The Si–C stretching vibration was found at 1167 cm−1 [36, 37]. The RHA was not showed these bands.
Catalysts synthesis using RHACCl as a precursor
L-Phenylalanine-Ru(III) complex immobilized onto RHACCl
L-(N-α-acetylphenylalanine)-Ruthenium(III) complex was immobilized onto silica in a simple procedure using RHACCl to produce a heterogeneous catalyst labeled as RHAPhe-Ru (Scheme 5) [38].
Figure 6a showed the 29Si MAS NMR spectrum and the discussion was adapted from [37]. At δ = –107.70 ppm the chemical shifts attributed to Q4 silicon atoms [9]. Q4 of RHACCl was deducted at chemical shifts (δ = -109.92 ppm), which was significantly shifted as compared with the RHAPhe-Ru. Single SiOH groups (silanol, Q3) on the catalyst were deducted at (-99.33 ppm). The Q3 of RHACCl was located at -100.56 ppm, which was found to shift slightly in comparison to the catalyst. A T3 chemical shift was found at -59.29 ppm indicating the formation of three siloxane bonds Si-O-Si. T3 of RHACCl was observed at -65.2. At -57.4 ppm the T2 of RHACCl was present which shifted to -55.42 ppm in RHAPhe-Ru.

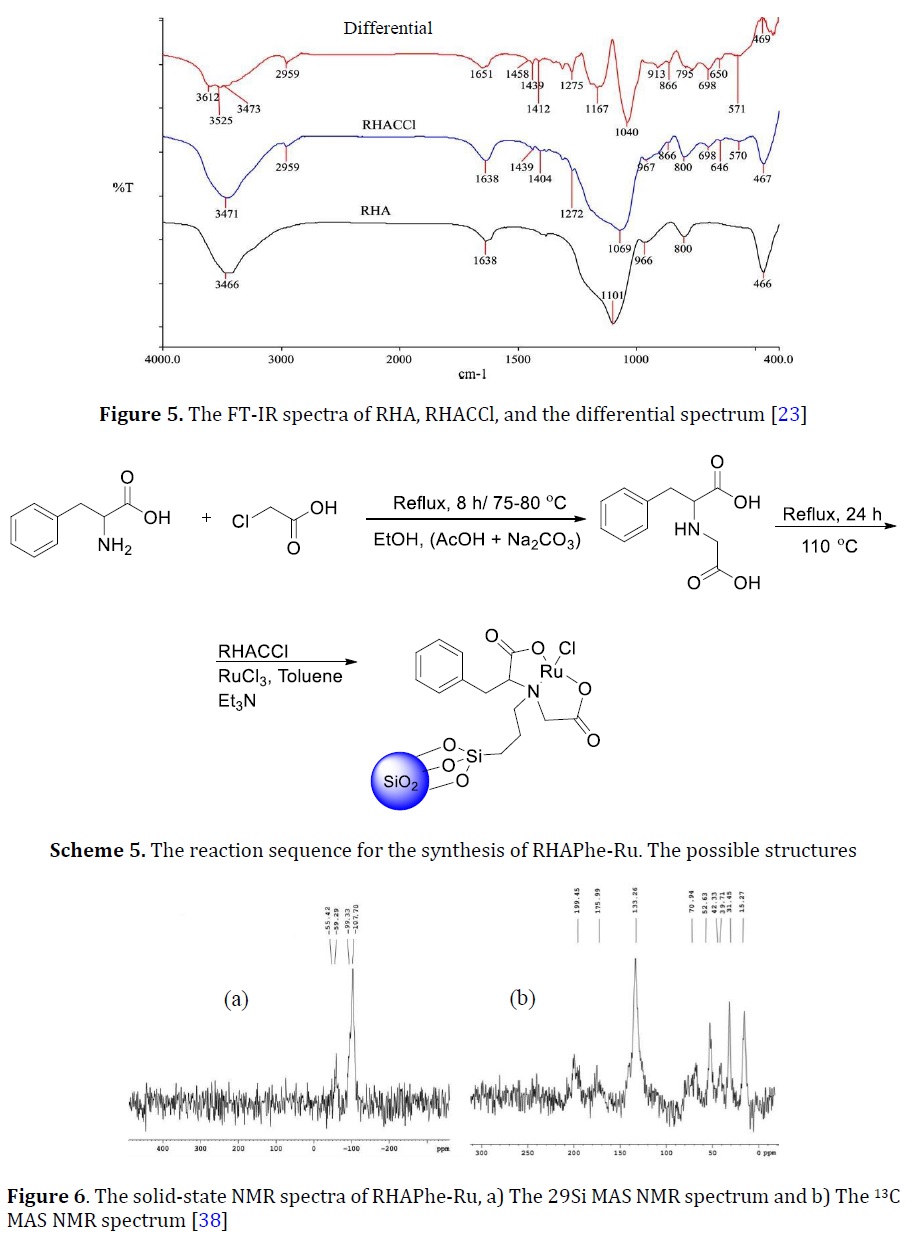
Figure 6b showed the 13C MAS NMR of RHAPhe-Ru [46]. Carbon directly connected to the silicon atom was observed at 15.72 ppm (Scheme 5). The C2 carbon was located at 31.45 ppm, while the terminal carbon atom bonded to the N was observed at 52.63 ppm. The RHACCl had high field chemical shifts for the propyl carbons compared to RHAPhe-Ru. Two strong chemical shifts at 39.71 and 42.33 ppm were assigned to the carbon libel at C8 and C4, respectively. Chiral carbon (C7) had a chemical shift at 70.94 ppm. The benzene ring had a broad chemical shift centred at 133.26. The carbonyl groups were observed at 175.99 and 199.45 ppm. The RHAPhe-Ru is used for the esterification rection of ethyl alcohol with acetic acid. About 82% of ethyl acetate was collected over the catalyst.
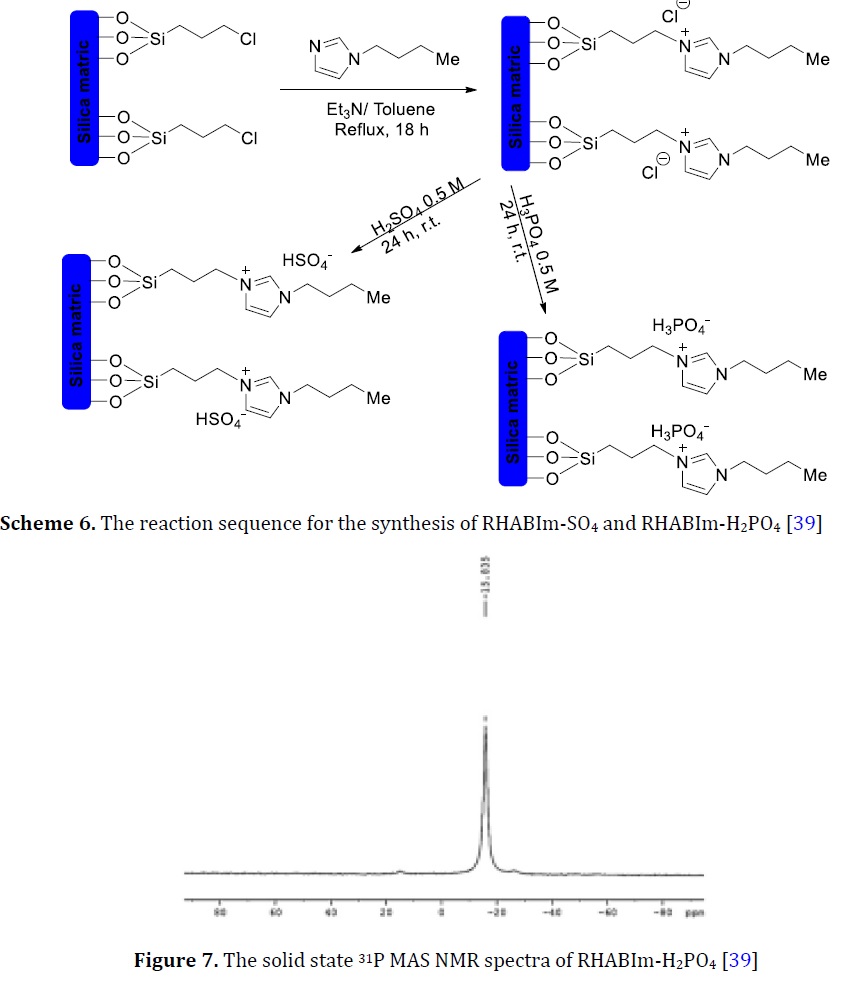
Preparation of N-heterocyclic carbene-silica via RHACCl
The terminal Cl in RHACCl was successfully replaced with 1-Butylimidazole RHABIm-Cl to produce quaternary amine [39]. Chloride of quaternary amine was sulfated and in a separate experiment phosphate was easily at room temperature. Each catalyst was labeled as RHABIm-SO4 and RHABIm-H2PO4, respectively (Scheme 6). RHABIm-H2PO4 was used to show the importance of the ionizable proton in acid catalysis. A single sharp chemical shift at -15.8 ppm was observed in 31P MAS NMR of RHABIm-H2PO4 (Figure 7, adapted from [39]). This peak proved the successful replacement of chloride ion by phosphate ions. The cyclization of glycerol to cyclic acetals was studded over RHABIm-SO4 and RHABIm-H2PO4. Six hours was recorded as the optimum time to produce 75-80% of five-member ring isomer over RHABIm-SO4 and RHABIm-H2PO4, respectively.
P-phenylenediamine and dithiooxamide immobilized onto RHACCl
The solid catalysts were prepared by the reaction of p-phenylenediamine (PDA) and dithiooxamide (DTO) silica-cl end group (RHACCl) to form RHAPDA and RHADTO, respectively [40]. About 145 m2gm−1 as a specific surface for RHAPDA was deducted. The RHADTO had 9.75 m2gm−1 specific surface area which is very low compared with RHAPDA. Both catalysts showed about 3.38 and 6.01% nitrogen in their structure as in elemental analysis. RHADTO showed 12.68% of sulfur in its structure.
The equation presented by Rahman et al. [41]. was used to calculated the percentage loading of PDA onto RHACCl and found to be 29.29%. The surface coverage by PDA was found to be 1.16 μmol m-2. Similarly, about 45.81% of DTA was grafting onto RHACCl. A surface coverage by DTA was calculated and found to be 1.77 μmol m-2. Both catalysts showed very promising activity for biodiesel production. Ethyl alcohol with acetic acid was used as a model esterification reaction for catalytic activity.
Silica-imidazole composite from RHACCl
Silica-imidazole (RHA-IMI) composite hexagonal structure was produced under reflexing by the immobilization of imidazole onto RHACCl [42].
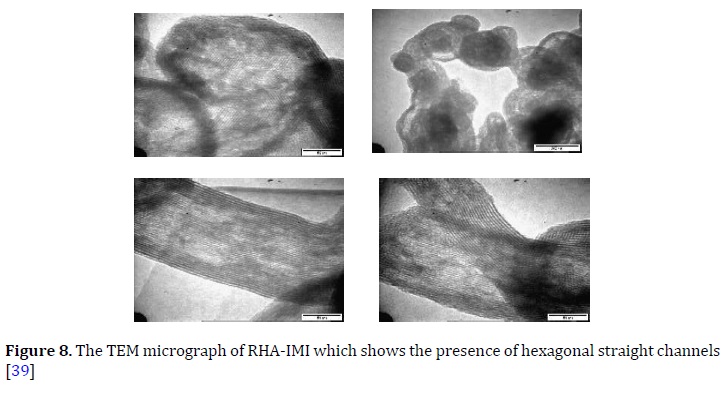
The hkl reflection planes of 100 and 110 were present at an angle at 2θ = 2.4° and 2θ = 3.2° in XRD [43, 44]. However, in the XRD pattern, the 200 and 210 for MCM-41 plane were not detected. The measuring size using the TEM measurement was ca. 2.8 nm. The TEM was conforming to the hexagonal straight channels and supported the XRD results (Figure 8) [50].
Preparation of 2,4-dinitrophenylhydrazanomethylphenol and salicylaldehydephenylhydrazone onto RHACCl
Salicylaldehydephenylhydrazone (Scheme 7a) and 2,4-dinitrophenylhydrazanomethylphenol (Scheme 7b) were immobilized onto silica via a simple method using RHACCl. The catalysts were labeled as RHPHMP and RHDNPH, respectively [45, 46]. The RHPHMP can efficiently hydrolyzed cellulose to its monomers, with higher glucose yield reaching 82% at 140 °C during 14 hours. The catalysts were repeated without the loss of catalytic activity during the cellulose hydrolysis. Homogenous RHDNPH was used for cellulose hydrolysis during 11 hours. Similarly, about 84% of glucose was formed with 100% selectivity.
Preparation of urea sulphate onto RHACCl
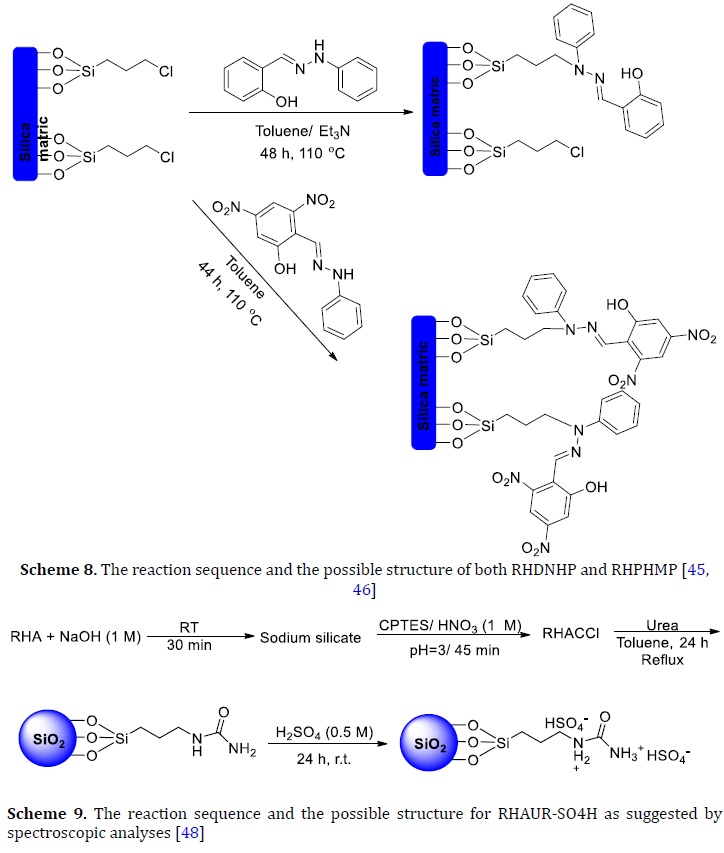
Urea successfully replaced the chloride end group onto RHACCl to prepare a heterogeneous catalyst. The amine end group onto the catalyst was treated with sulfuric acid to give quaternary ammonium denoted as RHAUR-SO4H (Scheme 9) [47].
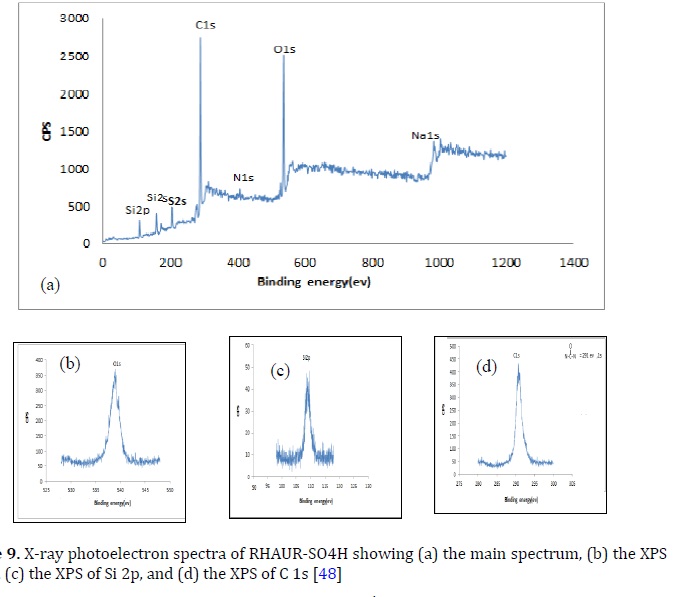
Figure 9 [48] showed the chemical analysis (XPS)of RHAUR-SO4H. The peaks of O 1s, Si 2p, S 2s, N 1s, and C 1s of the catalyst were presented in Figure 9. The O 1s binding energy was found to be532.3 eV for RHAUR-SO4H (Figure 9a and b), while the Si 2p binding energy was102.7 eV as seen in Figure 9a and c [48]. The C 1s binding energy of RHAUR-SO4H was found in the region of 291 eV, which can be seen in Figure 9a and d.
The spectral peak of S 2s XPS of RHAUR-SO4H appeared at ca. 168-169eV which bounded to sulfate (S 6+) species due to the –SO4H group. This finding was consistent with a prior study by González et al. [49]. The XPS (Figure 9a) also reveals a peak at ca. 1070 eV associated with Na 1 s species.
The Na presence is due to the synthesis process of the catalyst. The XPS spectra results proved that the urea was immobilized successfully onto silica, and then the amine groups were sulfated with sulfuric acid to give quaternary ammonium.
Cellulose was fully hydrolysis to glucose for 2 hours over RHAUR-SO4H. A mixture of cyclohexanol/LiCl was used as a reaction solvent. It was also found that after 7 hours the glucose formed was also hydrolysis to other products. The RHAUR-SO4H was easily recovered from the reaction mixture and reused several times with the same activity as in the fresh catalyst.
Silica functionalized 4-Aminophenazone
Polysulfone-based mixed-matrix membranes were prepared from polysulfone and mesoporous silica extracted from rice husk ash Gilan et al. [50]. The extracted silica was further functionalized with 4-aminophenazone via RHACCl to introduce polar amine CO2 philic groups to enhance the gas separation performance of membranes. The pure gas permeation data showed a simultaneous increase in permeability and selectivity of membranes with increased loading of functionalized silica. The enhanced solubility of CO2 on polar amine sites of the filler combined with its mesoporous large channels resulted in improved permeability and selectivity as compared with non-functionalized fillers.
Conclusion
This review provides a comprehensive view the latest methods to functionalize silica with CPTES to give RHACCl. The method was easily executed, greatly costly decreased, non-toxicity to the environment, effective, and speeding up the rate of producing large amounts of the catalysts. It was also discussed that different organic molecules with specific functional groups can be heterogenized onto silica via RHACCl to synthesize promising catalysts used in various reactions in industries and biological areas.
Acknowledgements
The Authors would like to thank Prof. Farook Adam (University Sains Malaysia, School of Chemical Science) for the valuable dissection and comments during the writing this review.
How to cite this manuscript: Antitubercular drugs: new drugs designed by molecular modifications. Asian Journal of Green Chemistry, 6(3) 2022, 370-378. DOI: 10.22034/ajgc.2022.4.6
)