Document Type : Original Research Article
Authors
- Shyamali Wijekoon 1
- Chinthika Gunasekara 2, 3
- Lalinda Palliyaguru 1
- Neluka Fernando 2
- Pradeep Jayaweera 1
- Upul Kumarasinghe 1
1 Department of Chemistry, University of Sri Jayewardenepura, Colombo, Sri Lanka
2 Department of Microbiology, University of Sri Jayewardenepura, Colombo, Sri Lanka
3 Center for Plant Materials and Herbal Products Research, University of Sri Jayewardenepura, Colombo, Sri Lanka
Abstract
A simple, efficient, and environmentally benign green approach was developed for the synthesis of 3-alkenyl oxindole derivatives with effective antifungal properties. The targeted compounds (3a-3g) were synthesized by using Knoevenagel condensation with oxindole and substituted aromatic aldehydes in the solvent-free, microwave-assisted reaction conditions. Silica extracted from rice husk waste was functionalized with (3-aminopropyl) triethoxysilane (APTES) to use as an eco-friendly, reusable, and solvent-free reaction medium. The structures of the final products were confirmed by their melting points, IR, 1H-NMR, 13C-NMR, and elemental analysis. The desired products were isolated with 72-88% yields in 12 minutes. The in vitro antimicrobial activity against ATCC standard cultures of human pathogenic Candida species, clinical isolates of Candida albicans, and several Gram-positive and Gram-negative bacteria were investigated and reported. All the synthesized 3-alkenyl oxindole derivatives exhibited the selective antifungal properties with the minimum inhibitory concentration (MIC) values ranging from 2 µg/mL to 125 µg/mL against tested Candida species. The findings of this study emphasized that green synthesized 3-alkenyl oxindole derivatives can be considered as the potential lead compounds to develop the effective antifungals against human pathogenic Candida species.
Graphical Abstract
)
Keywords
- Microwave
- Assisted Solvent
- Free 3
- Alkenyl Oxindole Derivatives Amino Functionalized Silica Antifungal Activity
Main Subjects
Introduction
Anti-microbial resistance (AMR) has been reported at an alarming rate, resulting in a significant global impact [1, 2]. The emergence of multidrug resistance among human pathogens has aggravated the challenges of managing these patients [3]. Candida albicans is an opportunistic pathogenic yeast and is a common cause of the fungal infections in humans [4, 5]. Moreover, the limited number of antifungal drugs available are becoming ineffective against these multi-drug resistant pathogens [3, 6]. As a result, there is an urgent need for the development of novel potential antifungal agents against resistant strains of fungal pathogens [7].
Oxindole scaffold is a ubiquitous pharmacophore found in various pharmaceutical and biologically active compounds [8⎼11]. The antimicrobial activity of oxindole derivatives has been investigated widely in recent years [12⎼15]. Among the reported oxindole derivatives, 3-alkenyl oxindole derivatives with an exocyclic C=C at C-3 position, are a remarkable group of biologically active compounds present in many natural products with the antimicrobial activity [16⎼19].
The synthesis of 3-alkenyl oxindole derivatives has been confirmed in the literature by the Knoevenagel condensation reaction (KCR) between the un-substituted oxindole and carbonyl compounds [20⎼23]. KCR necessarily requires the presence of different base catalysts, such as NaOH, diethyl amine, piperidine, and pyridine. This KCR has been reported as completed after 3-5 hours under the refluxing conditions in ethanol/methanol [20⎼25]. However, the synthesis of organic compounds by using an environmentally friendly green protocol has attracted much attention from researchers in last few years [26⎼31].
Microwave-assisted solvent-free reactions have many advantages, including improved atom-economy, E-factor, product purity, simplified recovery, and reusability, which can lead to environmentally friendly organic synthesis [32⎼34]. (3-aminopropyl) triethoxysilane (APTES) functionalized silica has been widely used in the solvent-free synthesis of biologically active organic compounds [35⎼38]. Furthermore, APTES-Silica has been identified as a cost-effective, environmentally friendly, efficient, and reusable adsorbent in recent publications [39-41].
This study reports a solvent-free microwave-assisted a novel green protocol for the synthesis of 3-alkenyl oxindole derivatives by using APTES functionalized silica as a solid reaction medium. The superior yield percentage (72%-88%) achieved here within 12 minutes of reaction time by using this green protocol has a considerable impact on the industrial application. In addition, the APTES-Silica recyclability increases the reaction sustainability. The silica used in this work was extracted from renewable rice husk waste. Furthermore, the antifungal activity of synthesized 3-alkenyl oxindole derivatives was investigated to assess their suitability as the potential lead compounds for the drug development in the future.
Experimental
Materials and methods
All the chemicals and reagents were purchased from Sigma Aldrich (USA) and they were used as received. The organic reactions were carried out in a 900 W microwave (Model No-SMW25GCQ6). Reactions were monitored by the aluminum-backed standard thin-layer chromatography (TLC) plates from Merck, Germany with fluorescent detection at 254 nm. Melting points of synthesized compounds were determined by using Melting point apparatus, DMP-400. The NMR spectra of synthesized 3-alkenyl oxindole derivatives were recorded by using Bruker Ascend 400 NMR spectrophotometer (Bruker, USA). Tetramethylsilane (TMS) was used as an internal reference and chemical shifts were reported as δ ppm units. Spectral data were presented as s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, and coupling constant (J) in Hertz (Hz). The IR spectra were recorded by Attenuated Total Reflection-Fourier Transform Infrared (ATR-FTIR) spectroscopy on Thermo Scientific Nicolet S10 FT-IR spectrometer. The UV-Visible absorbance was measured by using Thermo Scientific Multiskan Sky Microplate Spectrophotometer. SEM monographs of APTES functionalized silica was obtained in SEM(ZEISS)-EVO|LS15. EDX was performed in EDAX-elements. Likewise, Thermogravimetric Analyzer-TA SDT 650 was used to observe the characteristic weight losses of the APTES functionalized silica. CHN analysis was performed in Perkin-Elmer 2400 Series II CHNS/O Analyzer.
Microorganisms
The following microorganisms were obtained from the culture collection of Department of Microbiology, Faculty of Medical Sciences, University of Sri Jayewardenepura. Escherichia coli ATCC 25922, Staphylococcus aureus ATCC 25923, Pseudomonas aeruginosa ATCC 27853, Acinetobacter baumannii (clinical strain), and yeast cultures Candida albicans ATCC 10231, Candida glabrata ATCC 90030, Candida parapsilosis ATCC 22019, Candida krusei ATCC 6258, and three clinical isolates of Candida albicans. All bacterial cultures were maintained in Mueller Hinton Agar (MHA, Himedia, India) and fungal strains in Sabouraud’s Dextrose Agar (SDA, Himedia, India).
Methods
General procedure for synthesis of 3-alkenyl oxindole derivatives
Oxindole (1 mmol), aldehyde (1 mmol), and 0.5 g of APTES functionalized silica were added into a 25 mL round bottom flask and irradiated inside the microwave oven for 12 minutes. After completion of the reaction, the product was extracted into a mixture of ethyl acetate, acetone, and ethanol (1:1:1 v/v/v), concentrated in the rotary evaporator and purified by using silica gel column chromatography. Some products were purified by recrystallization from ethanol.
Preparation of APTES functionalized silica
Silica was extracted from rice husk following a sol-gel method by using microwave irradiation. Rice husk was thoroughly washed with distilled water before being treated with 3% (v/v) hydrochloric acid to remove any metallic impurities, and then the air-dried rice husk was calcined in a muffle furnace at 700 °C for 6 hours. The resulting ash was refluxed with 200 mL of 5 M NaOH under microwave irradiation for 5 minutes. The refluxed solution was filtered through Whatman no 1 filter paper and subsequently acidified (pH=3) with concentrated hydrochloric acid to obtain silica gel. The silica gel was filtered, dried, and grounded to obtain silica as a fine powder. This silica obtained from rice husk (10 g) was immersed in 100 mL of 6 M HCl and the mixture was refluxed for 5 hours. After the mixture was reached the room temperature, the solid was filtered and washed thoroughly with distilled water until acid-free. Subsequently, the product was dried in an oven at 100 °C for 48 hours to obtain activated silica [42]. Exactly 10 g of activated rice husk silica in 50 mL of toluene was added to a two-neck round bottom flask and refluxed at 100 °C, followed by gradual addition of 30 mmol of (3-aminopropyl) triethoxysilane (APTES) and refluxing was continued for further for 8 hours. The obtained solid was separated out by filtration, washed sequentially with toluene, acetone, water, and again with acetone twice (each 50 mL). The product was dried at 100 °C for 12 hours to obtain the APTES functionalized silica [43].
Antimicrobial activity
Thin-layer chromatography (TLC)-bio autography
Thin-layer chromatography-bio autography technique was carried out as described by Marston et al. with some modifications [44]. The TLC plate was sterilized by dipping it in 95% ethanol solution for 5 seconds. A dried TLC plate was used to separate the obtained compounds under sterile conditions. A small amount of oxindole derivative and the starting materials to be analyzed were spotted near the bottom of the TLC plate, and then the TLC plate was placed in the developing chamber containing 1:1 sterile hexane and ethyl acetate. The overnight incubated microbial cultures were used to prepare the standard microbial inoculum and the turbidity was adjusted to 0.5 McFarland standard in the sterile normal saline. Mueller Hinton agar plates were inoculated by spread plate method by using the standard inoculum (100 µL) to ensure even spreading. The developed TLC plate was placed face down on the inoculated Mueller Hinton Agar (MHA) plate and allowed to contact the agar surface for 30 minutes to enable the diffusion of separated compounds. The chromatogram was removed and subsequently, the agar plates were incubated at 37 °C for 24 hours. The inhibition zones on the agar surface corresponded to the spots in the chromatographic plate were observed and recorded.
Agar well diffusion assay
The overnight incubated cultures of bacteria in Mueller Hinton agar (MHA, Himedia, India) at 37 °C and yeast strains in Sabouraud’s dextrose agar (SDA, Himedia, India) at 25 °C were used to prepare the standard inocula in sterile normal saline by adjusting the turbidity to 0.5 McFarland standard. Standard inoculum (100 μL) of each organism was spread on MHA plates for bacteria and SDA plates for Candida species. Wells were prepared on the agar plates by using a 9 mm diameter sterile cork borer and the bottom of the wells was sealed with a drop of molten agar. Wells were loaded with 200 µL aliquots of the synthesized compounds dissolved in 40% propylene glycol (250 μg/mL). Gentamicin (10 μg) and vancomycin (30 μg) were used as the positive control for the selected bacteria and miconazole (30 μg) was used as the control drug for the antifungal activity. Solvent utilized to solubilize the compounds (propylene glycol 40%) was used as the negative control. Plates were incubated at 37 °C for 24 hours. All experiments were done in triplicate and average zones of inhibition (ZOI) was calculated [45].
Minimum Inhibitory concentration by Broth micro-dilution assay
The minimum inhibitory concentration (MIC) was determined according to the methods described in the literature with some modifications [46, 47]. Each test organism was cultured overnight at 25 °C on Sabouraud’s dextrose agar medium. Colonies were picked from the culture and suspension was prepared in sterile saline with turbidity adjusted to 0.5 McFarland scale corresponding to 1.5×108 CFU/mL. The suspension was diluted 1000 times to obtain the final inoculum which corresponds to 1.5×105 CFU/mL. The synthesized compounds were dissolved in Mueller Hinton Broth (MHB, Himedia, India) and double dilution series were prepared in MHB supplemented with 0.005% w/v Alamar Blue (Resazurin), and then 100 μL of diluted compound and 100 μL of microbial suspension were added to each well of a 96 well plate and incubated at 25 °C, aerobically for 48 to 72 hours. Following incubation, resazurin (blue and nonfluorescent) was reduced to resorufin (pink and fluorescent) in the presence of viable organisms. The minimum inhibitory concentration (MIC) for each tested microorganism was determined visually as the lowest concentration of the compound that resulted in blue color.
Minimum fungicidal concentration
The Minimum Fungicidal Concentration (MFC) of the synthesized compounds was determined according to the method described in the literature with some modifications [48, 49]. The synthesized compounds were dissolved in Mueller Hinton Broth (MHB, Himedia, India) and double dilution series were prepared in the same medium. Candida species suspension (1.5×105 CFU/mL) was added to each 96 well plates containing the synthesized compounds. After incubation at 37 °C for 48 to 72 hours, the MFC was determined by subculturing 5 μL aliquot from each microplate well on Mueller Hinton Agar (MHA, Himedia, India) plates. The plates were incubated at 37 °C and the fungal colonies grown were observed after 24-48 hours. The MFC values were determined as the lowest concentration of compounds where no colonies were visible on the agar plate. The assay was done in triplicate.
Spectroscopic data
(E) 3-(4-Methoxybenzylidene)indolin-2-one (3a)
Yellow solid, IR (KBr) (νmax/ cm-1): 3181 (NHCO), 3145, 3077, 3023 (CH), 2899 (CH), 1703 (NHCO), 1634, 1608 (CC), 1461(CC), 1327 (CNH), 1230, 1202, 1141, 692. The spectroscopic data were matched with those reported in the literature [25, 50]. 1H-NMR (400 MHz, CDCl3): δ 8.52 (s, 1H, NH-1), 7.78 (s, 1H, H-vinyl), 7.76 (d, J =7.6 Hz, 1H, H-4), 7.67 (d, J =8.4 Hz, 2H, H-2′,6′), 7.21 (dt, J = 0.80, 7.60 Hz, 1H, H-6), 7.00 (d, J =8.4 2H, H-3′,5′), 6.88-6.92 (m, 2H, H-5,7), 3.89 (s, 3H, OCH3-4′). 13C-NMR (100 MHz, CDCl3): δ 170.0 (s), 141.4 (s), 137.6 (s), 134.9 (s), 129.9 (s), 129.6 (s), 129.3 (s, 2C), 128.6 (s, 2C), 127.5 (s), 123.0 (s), 121.8 (s), 121.7 (s), 110.2 (s), 55.2 (s).
(E) 3-Benzylideneindolin-2-one (3b)
Yellow solid, IR (KBr) (νmax/ cm-1): 3181 (NHCO), 3153, 3078, 3024 (CH), 2890 (CH), 170 (NHCO), 1636, 1615 (CC), 1463 (CC), 1328 (CNH), 1232, 1204, 694. The spectroscopic data were matched with those reported in the literature [50]. 1H-NMR (400 MHz, CDCl3): δ 8.12 (s, 1H, NH-1), 7.84 (s, 1H, H-vinyl), 7.67-7.63 (m, 3H, H-2′,6′,4) 7.49-7.41 (m, 3H, H-3′,5′,4′), 7.21 (dt, J = 0.80, 7.60 Hz, 1H, H-6), 6.89 (d, J = 7.60 Hz, 1H, H-7), 6.85 (dt, J = 0.80, 7.60 Hz, 1H, H-5). 13C-NMR (100 MHz, CDCl3): δ 170.2 (s), 141.5 (s), 137.5 (s), 134.8 (s), 129.9 (s), 129.6 (s), 129.3 (s, 2C), 128.6 (s, 2C), 127.5 (s), 123.0 (s), 121.8 (s), 121.7 (s), 110.1 (s).
(E) 3-(3-Phenylallylidene)indolin-2-one (3c)
Orange solid, the spectroscopic data were matched with those reported in the literature [24, 51]. IR (KBr) (νmax/ cm-1): 3131 (NHCO), 3058 (CH), 2845 (CH), 1700 (NHCO), 1612 (CC), 1587, 1460 (CC), 1337 (CNH), 964, 734, 658. 1H-NMR (400 MHz, CDCl3): δ 8.00 (s, 1H, NH-1), 7.72 (d, J =7.60 Hz, 1H, H-4), 7.58-7.66 (dd, J=12.40, 12.00 Hz, 1H), 7.59 (d, 2H, J=7.20 Hz, 3′,5′), 7.49 (d, J =12.40 Hz, 1H), 7.44-7.35 (m, 3H, H-2′,6′,4′), 7.23 (dt, J =0.80, 7.60 Hz, 1H, H-6), 7.16 (d, J = 15.2 Hz, 1H), 7.07 (dt, J =0.80, 7.60 Hz, 1H, H-5), 6.90 (d, J =7.60 Hz 1H, H-7). 13C-NMR (100 MHz, CDCl3): δ 170.0 (s), 144.4 (s), 143.1 (s), 140.9 (s), 139.7 (s), 136.4 (s), 135.9 (s), 129.7 (s), 128.9 (s, 2C), 127.6 (s, 2C), 123.7 (s, 2C), 123.4 (s), 122.1 (s), 119.5 (s), 110.0 (s).
(E) 3-(4-Hydroxy-3-methoxybenzylidene) indolin-2-one (3d)
Yellow solid, IR (KBr) (νmax/ cm-1): 3383 (OH), 3164 (NHCO), 3063 (CH), 2850 (CH), 1682 (NHCO), 1618 (CC), 1574, 1504, 1470 (CC), 1338 (CNH), 786, 731; Anal. Calcd. for C16H13N1O3: C, 71.81; H, 4.90; N, 5.24; Found: C, 70.18; H, 4.71; N, 5.23. 1H-NMR (400 MHz, DMSO-d6): δ 10.52 (s, 1H, NH-1), 9.84 (s, 1H, OH-4′), 8.68 (s, 1H, H-vinyl), 7.74 (dd, J =2.00, 8.40 Hz, 1H, H-6′), 7.70 (s, 1H, H-2′ ) 7.65 (d, J = 7.60 Hz, 1H, H-4), 7.16 (t, J = 7.60 Hz, 1H, H-6), 6.97 (t, J = 7.60 1H, H-5), 6.87-6.81 (m, 2H, H-5′,7), 3.85 (s, 3H, OCH3-3′). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 167.9 (s), 150.2 (s), 147.4 (s), 140.4 (s), 138.3 (s), 128.3 (s), 128.3 (s), 126.5 (s), 126.1 (s), 123.3 (s), 121.2 (s), 119.4 (s), 116.3 (s), 115.6 (s), 109.5 (s), 55.9 (s).
(E) 3-(4-Hydroxybenzylidene)indolin-2-one (3e)
Yellow solid, IR (KBr) (νmax/ cm-1): 3159 (OH), 1677 (NHCO), 1603 (CC), 1580, 1510, 1462 (CC), 1279 (CNH), 1168, 775, 677. The spectroscopic data were matched with those reported in the literature [25]. 1H-NMR (400 MHz, DMSO-d6): δ 10.51 (s, 1H, NH-1), 10.12 (s, 1H, OH-4′), 7.69 (d, J = 7.60 Hz, 1H, H-4), 7.62 (d, J =8.4 Hz, 2H, H-2′,6′), 7.54 (s, 1H, H-vinyl), 7.21 (dt, J = 0.80, 7.60 Hz,1H, H-6), 6.93-6.86 (m, 4H, H-3′,5′,5,7). 13C-NMR (100 MHz, DMSO-d6): δ 169.6 (s), 159.7 (s), 143.0 (s), 137.0 (s), 132.2 (s, 2C), 129.8 (s), 125.5 (s), 125.0 (s), 122.4 (s), 121.7 (s), 121.5 (s), 116.1 (s, 2C), 110.4 (s).
(E) 3-(4-Dimethylaminobenzylidene)indolin-2-one (3f)
Orange solid, IR (KBr) (νmax/ cm-1): 3182 (NHCO), 3021 (CH), 2888 (CH), 1682 (NHCO), 1615 (CC), 1586, 1525, 1462 (CC), 1367, 1325 (CNH), 1094, 735, 675. The spectroscopic data were matched with those reported in the literature [25, 50]. 1H-NMR (400 MHz, CDCl3): δ 7.91 (d, J = 8.00 Hz, 1H, H-4), 7.82 (s, 1H, NH-1), 7.77 (s, 1H, H-vinyl), 7.68 (d, J = 8.80 Hz, 2H, H-2′,6′), 7.18 (t, J = 7.60 Hz, 1H, H-6), 6.92 (t, J= 7.60 Hz, 1H, H-5), 6.87 (t, J = 7.60 Hz, 1H, H-7) 6.76-6.72 (m, 2H, H-3′,4′). 13C-NMR (100 MHz, CDCl3): δ 170.6 (s), 151.5 (s), 140.7 (s), 139.2 (s), 134.8 (s), 132.2 (s, 2C), 128.4 (s), 127.0 (s), 122.3 (s), 121.5 (s), 118.1 (s), 111.2 (s, 2C), 40.6 (s, 2C),
(E) 3-[(Furan-2-yl)methylidenyl]indolin-2-one (3g)
Brown solid, IR (KBr) (νmax/ cm-1): 3486, 3137 (NHCO), 3012 (CH), 2890 (CH), 1690 (NHCO), 1608 (CC), 1473 (CC), 1358, 1323 (CNH), 741. The spectroscopic data were matched with those reported in the literature [25, 51, 52]. 1H NMR (400 MHz,CDCl3): δ 8.47 (d, J = 8.00 Hz, 1H, H-4), 7.97 (s, 1H, NH-1), 7.86 (s, 1H, H-vinyl), 7.67-7.63 (m, 3H, H-2′,6′,4) 7.49-7.41 (m, 3H, H-3′,5′,4′), 7.21 (dt, J = 0.80, 7.60 Hz, 1H, H-6), 6.89 (d, J = 7.60 Hz, 1H, H-7), 6.87 (dt, J = 0.80, 7.60 Hz, 1H, H-5). 13C-NMR (100 MHz, CDCl3): δ 170.4 (s), 151.3 (s), 145.9 (s), 140.9 (s), 129.5 (s), 128.7 (s), 125.2 (s), 122.2 (s), 120.7 (s), 120.2 (s), 119.2 (s), 113.2 (s), 109.7 (s),
Results and Discussion
Optimization of reaction conditions
The optimization of reaction conditions was done on the model system by using oxindole (1) and anisaldehyde (2a) as a starting material (Table 1). The optimal microwave irradiation time for obtaining the desired product (3a) was 12 minutes and gave a yield of 79 % (entry 4). There was no significant improvement in the yield after 12 minutes (entries 5 and 6). The highest conversion of starting materials was observed when using 0.5 g of APTES functionalized silica (entry 4). Only 21% yield of 3a was obtained when the reaction was performed under microwave irradiation in the presence of neat silica (entry 13). The reaction with APTES functionalized silica resulted in a trace amount of 3a at room temperature without microwave irradiation (entry 12). The control experiments showed that no product was formed in the absence of the reaction medium (entries 14 and 15). Relatively low yield of 3a was obtained when the reactions were performed with piperidine under microwave irradiation and the conventional heating at 140 °C in ethanol presence as a solvent (entries 16 and 17). Furthermore, only a trace amount of product was obtained when the reaction performed with APTES functionalized silica under the conventional heating at 140 °C for 12 hours. These results indicate that microwave-assisted solvent-free synthesis of 3-alkenyl oxindole derivatives in the presence of APTES functionalized silica is superior to the conventional methods.
To study the scope of the reaction medium, different types of aldehydes, ketones, and amides were reacted with the oxindole under the optimized reaction conditions. The obtained results indicate that only aldehydes give the expected products with sufficient yields (Table 2). Seven 3-alkenyl oxindole derivatives (3a-3g, Figure 1) were prepared by using the developed green protocol. The desired products were isolated with 72-88% yields in 12 minutes under solvent-free microwave irradiation (Table 2). A group of Japanese researchers established a solvent-free protocol for C-3 selective alkenylation of oxindole with aldehydes utilizing commercially available CeO2 as a heterogeneous catalyst which requires 10-15 hours of reaction time [22]. Gholamzadeh and co-workers have reported in completing the same reaction within 1 hour by using sulfonated silica [53]. However, the present study reports an efficient green protocol for the synthesis of 3-alkenyl oxindole derivatives with sufficient yields in 12 minutes of microwave reaction time.
Table 1. Optimization of the reaction conditions

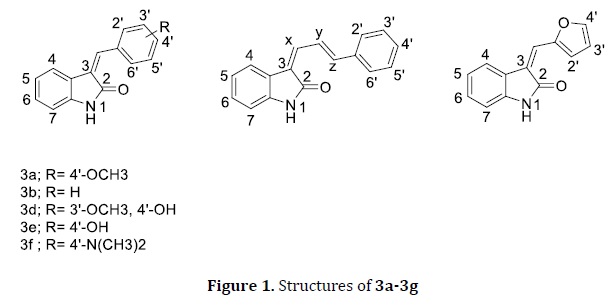

3-alkenyl oxindoles may exist as either E or Z isomers due to the exocyclic double bond presence at the C-3 position. The configurations of the synthesized compounds were assigned by comparison of chemical shifts of vinyl proton and aromatic protons (H-2´,6´) on the benzylidene ring [25, 50, 54]. Based on the previous reports, the chemical shifts of aromatic protons (H-2´,6´) on the benzylidene ring of E and Z isomers are in the range of 7.45-7.84 ppm and 7.85-8.53 ppm, respectively [24, 25]. The benzylidene ring protons of Z isomer being shielded by the carbonyl at C-2 position of the oxindole ring [55, 56].
In this study, all the synthesized compounds were obtained as predominant or single E isomeric form based on the 1H-NMR data. The E:Z ratios and the total yields of the products 3a-3g are listed in Table 2. The E/Z ratios were calculated by the integral height of H-2´,6´ protons and vinyl protons corresponding to E and Z isomers. The chemical shifts of H-2´,6´ protons of the predominant E and minor Z isomers of 3a, 3b, 3d, 3e, and 3f were found in the range of 7.61-7.75 ppm and 8.27-8.48 ppm, respectively. The chemical shift of vinyl proton in the compounds 3a, 3b, 3d, 3f, and 3g were found at 7.77-8.68 ppm and 7.47-7.60 ppm for the E and Z isomers, respectively. Furthermore, the chemical shift of alkyl proton (x) was used to assign the 3c configuration, which found at 7.68 ppm for the predominant E isomer while more downfield shift at 8.60 ppm was observed for the minor Z isomer. Based on the 1H-NMR spectral data, the product 3g was obtained as a single E isomeric form. The synthesized compounds obtained as predominantly or single E isomeric form may be due to the steric interference and electrostatic repulsion at the transition state [25]. The 1H-NMR spectra of 3d and 3e were obtained in DMSO-d6 due to their pure solubility in non-polar solvents. The spectra were recorded within two minutes after mixing with DMSO-d6 [51].
APTES functionalized silica
The combustion of rice husk waste within a muffle furnace produced 13% (w/w) rice husk ash. The silica in the ash was extracted as sodium silicate into the surrounding alkali solution under microwave irradiation for 5 minutes. In comparison, the silica extraction from rice husk ash following the conventional method requires more than 1 hour as reported previously [57]. This highlights the advantage of microwave method over conventional methods. Upon acidification of alkali sodium silicate mixture with concentrated hydrochloric acid a white color silica gel was precipitated (Figure 2). The percentage weight of the silica obtained from rice husk ash was 75% (w/w). Before functionalization with APTES, the trace metal impurities and hydrogen-bonded water molecules on the silica surface were removed via an activation process [58, 59]. The gross percentage weight (w/w) of the activated silica extracted from rice husk ash was 73%. This naturally occurring silica extracted from rice husk was used as a non-toxic, less expensive, and environmentally friendly silica precursor in the preparation of APTES functionalized silica [60, 61].
FT-IR analysis
The FT-IR spectra of neat rice husk silica and APTES functionalized rice husk silica are shown in Figure 3. The characteristic bands observed in the FT-IR spectra at 1080-1062 cm-1 and 796-792 cm-1 are due to the asymmetric and symmetric Si-O stretching vibrations in silica, respectively [53]. The absorption bands at 1633 cm-1 and 1630 cm-1 indicate H-O-H bending vibration of water. A broad band observed around 3100-3600 cm-1 is considered to be due to adsorbed water molecules [62]. The band at 951 cm-1 is attributed to the Si-OH stretching vibrations in silica [63]. The presence of -NH symmetric bending vibration bands were observed at 1570 m-1 in APTES functionalized silica. This indicates the successful functionalization of APTES onto the silica surface.


Thermogravimetric analysis
TGA thermograms of neat silica (a) and APTES functionalized Silica (b) are displayed in Figure 4. The TGA profiles of both neat silica and APTES functionalized silica gives two abrupt weight losses. The first one is at 100 °C is due to evaporation of the absorbed water, which is completed between 150 °C and 200 °C. Alkyl groups of functionalized silica brings more hydrophobicity leading to less water vapor adsorption and less weight loss compared with the unmodified silica [64]. The second weight loss starts above 200 °C and completes 500-600 °C is attributed to leave strained siloxane groups due to condensation of vicinal hydroxyl groups of silica samples [65].
PXRD patterns
Powder X-ray diffraction patterns of neat rice husk silica and APTES functionalized silica are depicted in Figure 5. The PXRD patterns of both types of silica show a hump centered at 2 = 22.0°, which matches perfectly with the standard Joint Committee on Powder Diffraction Standards (JCPDS) Card No. 29-85 of amorphous silica [62, 66, 67].
SEM/ EDX analysis
The morphology of neat silica extracted from rice husk waste is demonstrated in Figures 6a (´500 magnification) and 6b (´10 000 magnification). The scanning electron microscopic images revealed the polygonal morphology of the obtained silica. During the activation process, the neat silica resulted in micelle formation which was further aggregated to form siloxane (Si-O-Si) network (Figure 6c). This aggregated polymer network facilitates amine functionalization on the surface of silica [68, 69]. The SEM image of APTES functionalized silica exhibited near spherical morphology as observed in Figure 6d. The SEM/EDX analysis of activated silica (Figure 6e) and APTES-Silica (Figure 6f) confirmed the modification of silica with APTES, where nitrogen and carbon can be detected only in APTES-Silica in addition to silicon and oxygen [70].



CHN analysis
The CHN analysis provided the percentage of carbon, hydrogen, and nitrogen in the APTES functionalized silica. The percentage values of C, H, and N were obtained as 3.47, 0.93, and 1.17 (wt.%), respectively. The experimental C/N ratio of 3.46 of APTES functionalized silica, compared with the theoretical value of 3.00, indicates that almost all the ethoxy groups in APTES condensed with the hydroxyl groups of silica [71]. The surface coverage of functionalized silica was calculated by using N% since the only nitrogen source is APTES. The calculated surface coverage is 0.84 mmol/g (Supplementary information, Table S1).
Proposed reaction mechanism
The plausible reaction mechanism for the condensation reaction of oxindole (1) and aldehyde (2) catalyzed by APTES-silica is illustrated in Figure 7. The reaction initiates by deprotonation of the activated methylene group at C-3 position of the oxindole ring by NH2 group of the APTES-silica to form enolate II. In the second step, the enolate II acts as a nucleophile to attack on carbonyl carbon of the aldehyde 2. The aldol intermediate product III is protonated by regenerating amine-functionalized silica in the third step. Finally, 3-alkenyl oxindole derivative (3) is obtained upon the dehydration of intermediate product V.
Reusability of the reaction medium
The reusability of the reaction medium was evaluated by using the reaction of oxindole (1) and anisaldehyde (2a) with the optimized reaction conditions. After completion of the reaction, the reaction mixture was dissolved in a mixture of solvents (ethyl acetate, ethanol, and acetone), and the APTES-silica was separated by suction filtration, washed with ethanol twice, and dried at 100 °C for 3 hours. The recovered reaction medium was used for five consecutive runs under similar reaction conditions. The yield decreases by 27% in the regenerated samples after five cycles (Figure 8). These findings confirmed the published data on environmentally friendly and reusable APTES functionalized silica [39-41]. The environmental friendliness of the reaction has been further improved in the present study by using naturally occurring silica extracted from rice husk for the preparation of the solid reaction medium.


Biological activities
Thin layer chromatography (TLC)-bioautography for antimicrobial screening
Thin-layer chromatography (TLC)-bioautography technique was used as a simple qualitative detection method for the preliminary antimicrobial screening of the synthesized oxindole derivatives. The sterile dried chromatogram was obtained after the separation of starting materials: oxindole (2a) anisaldehyde (2b) and the products (2c) are indicated in (Figure 9A). The bioautography was carried out for Candida albicans, ATCC 10231 (Figure 9 I) and Escherichia coli, ATCC 25922 (Figure 9 II). Areas of growth inhibition were observed for both aldehyde (2b) and the product (2c) indicating the potential antifungal activity. No growth inhibition was detected against E. coli.

Agar well diffusion assay
The potential antimicrobial activity of the products was determined by using well diffusion assay against the selected microbial species as presented in Table 3. All the compounds exhibited a Zone of Inhibition (ZOI) against all tested Candida species (Figure 10). Compounds 3a, 3b, and 3c demonstrated the larger inhibition zones compared with other oxindole derivatives synthesized in the study. Results of well diffusion assay against S. aureus and MRSA indicated ZOIs of 12.33±0.23 to 13.66±0.47, while Gram-negative pathogens P. aeruginosa, A. baumanii, and E. coli did not show any inhibition zones at the tested concentrations. The Vancomycin control had ZOI against S. aureus (18.66±0.23) and MRSA (18.66±0.47). Gentamycin showed ZOI against P. aeruginosa (13.66±0.23), A. baumanii (13.33±0.23), and E. coli (18.66±0.23). The cell wall structure of Gram-negative bacteria may act as a permeability barrier for the tested compounds resulting in the poor antimicrobial activity [72].


Minimum Inhibitory Concentration (MIC)/Minimum Fungicidal Concentration (MFC)
Minimum Inhibitory Concentrations were determined visually based on color change due to the reduction of resazurin dye from blue to pink by viable organisms (Table 4, Figure 11). The solvent, propylene glycol 40 %, did not show any growth inhibition against tested organisms. All seven synthesized compounds (3a to 3g) exhibited a range of MIC/MFC values from 2 µg/mL to 63 µg/mL against tested Candida ATCC cultures, while the C. albicans clinical isolates demonstrated a higher MFC range (8-125 µg/mL, Table 4). The compound 3b revealed the lowest MIC values (2-8 µg/mL) and MFC values (2-16 µg/mL) against the four ATCC Candida species and the tested clinical isolates (8-16 µg/mL). Compound 3a had the second lowest MIC/MFC values of 8-32 µg/mL against four standard Candida species. The MIC and MFC values of 3a against tested clinical isolates were 16-32 µg/mL and 16-63 µg/mL, respectively. In the presence of compound 3b, Candida albicans ATCC10231 was found to have the lowest MIC/MFC value (2 µg/mL), while Candida glabrata and Candida krusei both had MIC value of 4 µg/mL. Candida parapsilosis had a MIC value of (8 µg/mL) when tested against compound 3b. The lowest MFC values of compound 3b against Candida glabrata, Candida krusei, and Candida parapsilosis were 8 µg/mL, 16 µg/mL, and 4 µg/mL, respectively. The MIC/MFC values of the antifungal drug miconazole against tested Candida species were £ 2 mg/mL.


According to the results of the MIC/MFC, 3-alkenyl oxindole derivatives were found to have the potent antifungal properties towards Candida species. Compound 3b with an unsubstituted phenyl group at C-4’ position had a higher antifungal activity. Compound 3a with methoxy substituted phenyl group had the second highest antifungal activity. Other compounds with hydroxy substituted phenyl ring in 3e, both methoxy and hydroxy substituted phenyl ring in 3d and N,N-dimethyl amino substituted phenyl ring in 3f were found to have the milder activity. Furthermore, the fural ring substituted compound 3g also demonstrated mild antifungal activity. Surprisingly, compound 3b which showed the highest antifungal activity, is similar to the antifungal drug miconazole against the standard culture of Candida albicans (ATCC10231).
Strigacova et al. reported oxindole derivatives with an exocyclic double bond at C-3 position exhibited the potential antifungal activities [11] which supports the fungicidal activity of synthesized 3-alkenyl oxindole derivatives. A series of triazole derivatives of 2-oxindole nucleus with exocyclic C=C have been synthesized and found to exhibit superior antifungal activity against C. albicans [73]. Furthermore, the antifungal potential of 3-alkenyl oxindoles bearing 3-alkenyl moiety has been investigated by Olgen et al. in Turkey. They reported the antifungal behavior of these derivatives dissolved in RPMI 1640 against two groups of human pathogenic candida species (C. albicans and C. krusei). However, the prominent antimicrobial activity was not observed due to the poor solubility of these derivatives in an aqueous solvent [74]. This is the only study found in the literature reporting the antifungal activity of 3-alkenyl oxindole derivatives. Therefore, in the present study propylene glycol, a pharmaceutical cosolvent [75] was used to dissolve the synthesized oxindole derivatives to evaluate the antifungal activity.
The discovery of the novel antifungal drug candidates is important as there is a limited number of antifungal drugs currently available for the clinical use. The findings of this study suggest 3-alkenyl oxindole derivatives as a potential antifungal candidate for further investigations.
Conclusion
In conclusion, the authors developed an environmentally friendly and efficient method for the synthesis of 3-alkenyl oxindole derivatives. In this green protocol, APTES functionalized silica was used as a reusable, solvent-free reaction medium to proceed the microwave-assisted reaction. The antimicrobial potential of the synthesized oxindole analogs was evaluated against a wide range of human pathogenic microorganisms. Compounds 3b, 3a, and 3c were found to have the potential antifungal activities against Candida species, among them 3b with an unsubstituted phenyl ring emerged as the most potent antifungal agent. These findings will endow with a great impact on organic chemists and medicinal chemists for assisting investigations on the development of the potential antimicrobial agents in the future.
Acknowledgments
The authors would like to thank the following organizations and people assisted to complete this work: Central Instrument Center (Faculty of Applied Sciences, University of Sri Jayewardenepura); Ms. L.B. Achala E Bogahawatta, Ms.Shamini Wickrama, (Department of Microbiology, Faculty of Medical Sciences, University of Sri Jayewardenepura); Sri Lanka Institute of Nanotechnology (SLINTEC).
Disclosure Statement
No potential conflict of interest was reported by the authors.
Funding
This work was financially supported by a university research grant ASP/01/RE/SCI/2017/81 (University of Sri Jayewardenepura).
Authors' contributions
All authors contributed to data analysis, drafting, and revising of the paper and agreed to be responsible for all the aspects of this work.
Orcid
Shyamali Wijekoon 0000000301462058
Chinthika Gunasekara 0000-0002-6003-3088
Lalinda Palliyaguru 0000-0003-4170-8534
Neluka Fernando 0000-0001-7551-8224
Pradeep Jayaweera 0000-0001-9263-1480
Upul Kumarasinghe 0000-0003-1975-8949
Supporting Information
Copies of 1H-NMR (400 MHz, CDCl3), and 13C-NMR (100 MHz, CDCl3) spectra of synthesized compounds and calculation of C/N ratio and APTES coverage based on CHN analysis (PDF).
How to cite this manuscript: Shyamali Wijekoon, Chinthika Gunasekara, Lalinda Palliyaguru, Neluka Fernando, Pradeep Jayaweera, Upul Kumarasinghe*. Solvent-free synthesis and antifungal activity of 3-alkenyl oxindole derivatives. Asian Journal of Green Chemistry, 6(4) 2022, 297-319. DOI: 10.22034/ajgc.2022.4.2
)