Document Type : Review Article
Authors
1 Glocal School of Pharmacy, Glocal University, Mirzapur Pole, Saharanpur, Uttar Pradesh, India
2 Laboratory Medicine Department, Faculty of Applied Medical Sciences, Umm Al‒Qura University, Makkah, 21955, Saudi Arabia
Abstract
Tuberculosis is a long-term infection caused by the bacteria Mycobacterium tuberculosis. Long-term TB treatment has several drawbacks, including medication resistance, noncompliance with therapy, and a scarcity of therapeutic options. It is critical to find new medications that are both safer and more effective. Molecular modification is a valuable method for identifying novel chemicals that can overcome these obstacles. The primary tactics to obtain a novel anti-tubercular medication are summarized in this paper.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
Tuberculosis (TB) is a chronic infectious illness that is also known as the "great white plague" owing to Mycobacterium tuberculosis (Mtb) infection. Mtb is an aerobic bacillus with high lipid content in its cell wall, resulting in high lipophilicity and resistance to alcohol, acids, alkali, and certain disinfectants. TB is the greatest cause of death worldwide. Every minute, one person in a poor country dies of TB. According to the World Health Organization (WHO), about one-third of the world's population is afflicted with tuberculosis. One billion people will be newly infected by 2020, over 125 million will become unwell, and over 30 million would die from TB if it is not managed. One of the most challenging aspects of treating TB is detecting and curing a large enough number of patients to stop transmission. The high rate of the Mtb infectivity is epidemiologically characterized, therefore one-third of the latent infection population that remains a reservoir for mycobacterium is the biggest impediment to TB control [1⎼5].
Mtb has a complex cell wall made up of mycolic acids (long-chain fatty acids with 60 to 90 carbon atoms), glycolipids, peptidoglycan, and proteins. TB is spread by the coughing of a sick person. The inhaled droplets carrying expectorated Mtb reach the host lung, starting the infection process [6]. When Mtb infects the human lung, it triggers an inflammatory response involving macrophage activation, which results in the release of pro-inflammatory cytokines such as interleukin 6, IL12, IL1B, and INF-γ. Mcl1 antiapoptotic protein is produced by Mtb-infected alveolar macrophages and is important for interrupting apoptosis in infected macrophages by regulating the mitochondrial membrane and blocking the release of cytochrome C and DNA-degrading enzymes [7⎼9]. The immune response fights Mtb, allowing the additional cells to be attracted to the affected side to stop the bacteria from growing and forming granulomas. The granuloma is an ordered structure created by immune cells in response to an antigenic stimulus that can be seen in both latent and active TB. The bacillus finds a home in the granuloma. There is a decrease in immune response activity metabolism, resulting in a condition of quiescence. The latent TB is a kind of Mtb dormancy that, unlike active TB, is not considered as an infectious illness [10]. Immunosuppressive circumstances reduce the immune system's effectiveness, allowing formerly dormant bacteria to reactivate, resulting in active TB decades after the initial infection. The bacteria may exist in balance with an immune response in normal circumstances, but in situations such as genetic disability, intercurrent illnesses (AIDS), starvation, and drug treatments, an imbalance can arise, and the Mtb multiplies rapidly, leading to TB [11]. When compared with merely TB-infected persons, the probability of developing active TB in a co-infected individual with HIV is 100 times higher [12]. Atypical mycobacteria such as Mycobacterium avium, M. kansassi, M. fortuitum, and M. chelonae are more prone to infections in AIDS patients. Furthermore, TB promotes AIDS development in HIV-positive individuals by increasing cytokine production and decreasing CD4+T cell numbers [6].
Multidrug-resistant tuberculosis (MDR-TB) is another major issue in the global fight against TB. Patients who had never been treated with an anti-TB drug showed resistance. MDR-TB causes around 460 thousand new cases every year, with about 740 thousand new patients infected with both Mtb and HIV/AIDS. About 10% of all new TB infections are resistant to at least one anti-TB drug because monotherapy contributed to the resistance development, genuine anti-TB drug therapy has included the use of numerous drugs [13]. Short-course chemotherapy (DOTs) consists of taking isoniazid (INH) and rifampicin (RIF) for six months, with pyrazinamide (PZ) and ethambutol (EMB) supplementation for the first two months [14]. MDR-TB and extensively drug-resistant (XDR) TB have arisen as serious therapeutic challenges. MDRTB is a kind of TB produced by bacteria that are resistant to or do not react to at least INH and RIF. Inappropriate therapy is the major cause of MDR-TB.
The improper use of anti-TB drugs or the use of low-quality medicines is the cause of drug resistance. MDR-TB can be treated and cured using the second-line drugs. When first-line drugs fail, these drugs are utilized. On the other hand, these drugs are poisonous and have serious adverse effects. The extensive therapy (often used for two years and need daily injections) is more costly and can cause major adverse drug responses in patients. MDR-TB is a subtype of XDR-TB and resistant to nearly all anti-TB drugs, including the two most effective first-line drugs, isoniazid (INH) and rifampicin (RIF). The best second-line anti-TB drugs are fluoroquinolones (FQs), and at least one of three injectable drugs (amikacin, kanamycin, or capreomycin). Patients are left with therapy alternatives that are less effective and typically have worse treatment results since XDR-TB is resistant to the most first- and second-line drugs. Persons with HIV infection or the other disorders impairing the immune system should be especially concerned about XDR-TB. Once infected, these individuals are more likely to acquire TB and have more chance of mortality. In addition, it is estimated that 9.6% of MDR-TB patients have XDR-TB [15⎼18].
Drugs currently in use to treat tuberculosis
The first-line drugs like isoniazid (INH) (together with pyridoxal phosphate to prevent peripheral neuropathy created by INH), ethambutol (EMB), pyrazinamide (PZ), and rifampicin (RIF) are used for two months, followed by INH and RIF alone for another four months. A weekly, directly monitored RIF regimen with INH for three months was shown to be equally effective as a daily regimen for preventing active TB in HIV-negative people with latent TB. The RIF-INH regimen showed greater treatment completion rates and lower hepatotoxicity rates. The reason for the prolonged treatment is that eradicating the infection from the body is extremely tough. After six months, the patient is declared cured (although there is still a relapse rate of 2 to 3 percent). The typical therapy for the latent TB is six to nine months of INH alone. These drugs are effective, have low toxicity, and are well tolerated by patients. These drugs are very useful for those who have active TB illness and have never had TB therapy. However, due to the high prevalence of resistance, streptomycin (STR) is no longer used as a first-line anti-TB drug. The second-line drugs are considered as a reserved therapy for the TB treatment [17⎼19].
When patients are resistant to the first-line drugs, XDR‒TB or MDR‒TB arises, and the second-line drugs are applied for the TB treatment. A drug may be classed as a second-line drug instead of the first-line drug for one of three possible reasons: it may be less effective than the first-line drugs, it may have toxic side effects or it may be unavailable in many developing countries. For MDR-TB and XDR-TB used the blend of the first-line drugs and second-line drugs like aminoglycosides (amikacin and kanamycin), polypeptides (capreomycin, viomycin, envyomicin), FQs (ofloxacin, levofloxacin, moxifloxacin, and gatifloxacin), thioamides (ethionamide and prothionmide), cycloserine (CYC), terizidone, and para-aminosalicylic acid (PASA). This chemotherapy is less effective, long, expensive, and more toxic than short-course therapy [14]. The third-line drugs include rifabutin, thioridazine, macrolides (clarithromycin), linezolid, thiacetazone, arginine, and vitamin D are still being developed, have less or unproven efficacy, and are very expensive [20].
Multidrug‒Resistant tuberculosis (MDR‒TB)
Drug resistance in M. tuberculosis arises in two ways:
1) MDR-TB requires resistance to at least two first-line anti-TB drugs, INH and RIF;
2) XDR-TB requires resistance to both the first- and the second-line anti-TB drugs [21].
Several biochemical steps are necessary for Mtb resistance, including:
- a) A reduction in the intracellular level of the drug by modifying the permeability of outer and inner membranes;
- b) Mutation or change of the cellular target; and
- c) The drug’s action on a diverse target [22, 23].
Mutations in genes liable for encoding enzymes accepted as targets are the core factor to produce resistance to anti-TB drugs. The exact interaction of RIF with the β‒subunit of RNA polymerase inhibited transcription and caused cell death. Mutations in the rpoB gene, which encodes the chain β enzyme, produce drug resistance by reducing the interaction of RIF with the polymerase [24]. The INH requires the katG gene product for its activation; INH is active after metabolism of Mtb catalase-peroxidase inhibiting the enoyl-ACP reductase, since it is encoded by the gene; its resistance involves four genes:
- a) katG, encodes the catalase-peroxidase;
- b) inhA in mycolic acid biosynthesis;
- c) ahpC, encodes alkyl hydroperoxide reductase C; and
- d) oxyR, regulator of oxidative stress [25, 26].
Isoniazid (INH) targets are KatG and inhA genes. KatG gene encodes two enzymes catalase/peroxidase that activates INH and peroxynitrite concerned in pathways of reactive nitrogen and oxygen intermediates. InhA gene encodes NADH-dependent enoyl-Acyl Carrier Protein (ACP)-reductase that inhibited the mycolic acid synthesis [27⎼29].
Ethambutol (EMB) resistance is given by mutations in embA, embB, and eembC genes that encode enzymes involved in arabinano synthesis [30]. Arabinosyl transferase is a target for the EMB action in both Mtb and M. avium. The enzyme is encoded by the embCAB gene organized as an operon and engaged in arabinogalactan synthesis [31].
Rifampicin (RIF) is still used as the best choice of the anti-TB drug. RIF is lipophilic and diffuses across the cell membrane of Mtb. It mainly targeted rpoB of DNA dependent-RNA polymerase β-subunit and rpoB uses four ribonucleotide triphosphates as substrates to catalyze DNA transcription into mRNA. RIF binds to the β-subunit of DNA-dependent-RNA polymerase and inhibits mycobacteria transcription [32⎼35].
Pyrazinamide (PZ) is inhibiting the fatty acid synthesis of Mtb. Its resistance occurs by mutations in pncA gene, encoding pirazianamidazes, that hydrolyzes the drug to make it active [36]. PZA is activated to pyrazine acid by the pyrazinamide/nicotinamidase encoded by gene pncA. Acidic condition favors the progress of protonated pyrazinoic acid passing through a membrane and is collected in the cell membrane of Mtb which interrupts cell membrane potential and changes membrane transport [37, 38]. RpsA gene encodes 30S ribosomal protein S1 accountable for the mRNA translation. Gene panD contributes to pantothenate biosynthesis by converting L-aspartate into β-alanine. PZA new target, clpC1 (Rv3596c) encodes an ATP‒dependent ATPase that is accountable for protein degradation by complex formation with protease clpP1 and clpP2 [39⎼41].
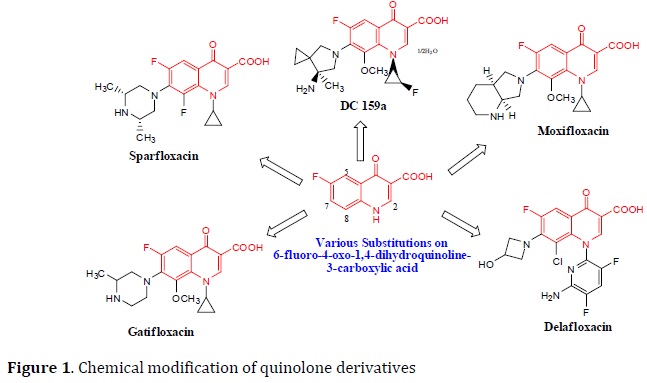
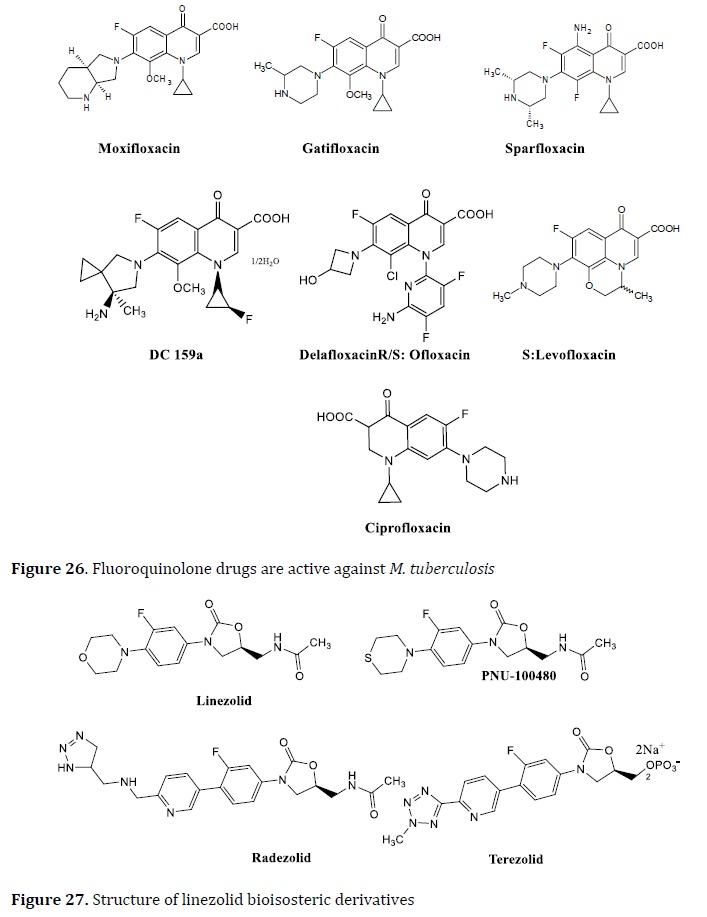
Fluoroquinolones (ciprofloxacin, levofloxacin, moxifloxacin, and gatifloxacin) (Figure 1) are mainly dependent on the blocking of mycobacterial DNA replication by binding to α and β subunits of DNA gyrase (gyrA and gyrB), which catalyze the DNA supercoiling, and finally inhibits DNA synthesis [42, 43].
Streptomycin is the first antibiotic used to cure TB. It targets both rpsl and rrs genes that encode 30S ribosomal protein S12 and 16S rRNA, respectively, and inhibits the translation initiation in the protein synthesis [44, 45].
Capreomycin resistance is caused by a mutation in the thyA gene [46]. The ethioamine is a mycolic acid inhibitor that inhibits cell wall synthesis due to the mutations in genes inhA (involved in mycolic acid synthesis) and ethA, encoding monooxygenase flavin, which is accountable for activating ethioamine [26].
Para-aminosalicylic acid resistance is due to mutations in thyA genes, which encode the synthase and regulates intracellular levels of folate [47]. PAS inhibits folic acid synthesis by the dihydropteroate synthase and dihydrofolate synthase effect that generates hydroxyl dihydrofolate antimetabolite inhibiting dihydrofolate reductase [48]. Secondly, inhibits cell wall parts mycobactin synthesis by reducing the iron uptake [33].
Ethionamide, two genes that take part in the role of ethA and inhA. EthA is regulated by the transcriptional repressor ethR [49]. Ethionamide disrupts mycolic acid synthesis by which monooxygenase-activated ethionamide binds to NAD+ and forms adduct inhibiting enoyl acyl‒ACP reductase enzyme [50⎼53].
Cycloserine is an analog of serine hydroxamic acid and terizidone. Its acts by interfering with mycobacterial cell wall synthesis by inhibition of L-alanine racemase encoded by alrA that forms D-alanine from L-alanine and D-phenylalanine synthetase enzyme that is essential for the peptidoglycan and cell wall synthesis by the inclusion of Dalanine into pentapeptide [54, 55].
Aminoglycosides (kanamycin and amikacin) and polypeptides (capreomycin and viomyocin) inhibit protein synthesis. Kanamycin and amikacin alter 16S rRNA and capreomycin and viomycin interfere with subunits of the 70S ribosome [56⎼58].
Linezolid is an oxazolidinone derivative interrupting protein synthesis by binding to the assembly of the 23S ribosomal RNA of the 50S subunit. The gene rplC and rrl are involved in the action. The rplC gene possesses 654 bp in length encoding 50S ribosomal L3 protein to give to the synthesis of ribosomal peptidyltransferase. Hence, rrl gene possesses 3138 bp length that encodes 23S ribosomal RNA [59].
Newer TB Drugs Against MDR‒TB
Bedaquiline or TMC207 is a diarylquinoline derivative and bactericidal. TMC207 involves blocking the proton pump of the ATP synthase of Mtb, then reducing the energy demand of mycobacteria and causing cell death [60, 61].
Delamanid or OPC 67683is a dihydro-nitroimidazo-oxazole derivative and is activated by the deazaflavin-dependent nitroreductase enzyme (Rv3547). It interrupts the mycobacterial cell wall component synthesis. It also inhibited the methoxy- and keto-mycolic acid synthesis which is a part of the mycobacterial cell wall. It is effective against mycobacterium [62, 63].
PA‒824 is a nitroimidazole derivative and activated by deazaflavin-dependent nitroreductase as the delamanid. The mechanism action of PA‒824 is not clear, but it could be explained as activity in aerobically replicating mycobacterial cell PA‒824 disrupts mycolic acid synthesis by the collection of hydroxy‒mycolates instead of ketomycolates [64, 65]. Accordingly, in hypoxic nonreplicating mycobacteria, PA‒824 releases Nitric oxide (NO) that obstruct cytochrome oxidase to disturb the energy metabolism of the cell wall [66, 67].
SQ‒109 is an EMB derivative, the mechanism is not clear and has no inhibition effect against secreted Ag85 mycolyltransferase. Rather SQ‒109 causes the gathering of trehalose monomycolate a precursor of trehalose dimycolate by delaying assembles of mycolic acids into the bacterial cell wall. Mmpl3 is a target of SQ‒109 and a transporter of trehalose monomycolate of the mycobacteria [68].
Molecular Modification
Molecular modification (MM) is a chemical alteration in a molecule that could be a lead compound aiming to enhance pharmacokinetics or pharmacodynamics. This strategy is used to discover many available drugs. Among MM, are the prodrug approach, molecular hybridization, and bioisosterism. Each one of these strategies will be prominent in its application in drug discovery [69]. Among them, molecular hybridization is a leading one in developing suitable lead molecules for the target disease. The "one‒target‒one‒drug" paradigm has been designed for one drug for a single target, many diseases remain inadequately treated today. This approach fails to treat some diseases, the drug discovery explores the hybridization between molecules to modulate multiple targets [70]. Nowadays, various multiple target therapy methods are used mainly in the conditions of unresponsive patient such as:
- a) Use of two or more individual formulations;
- b) Use of fixed-dose combination therapy in which two or more drugs are combined in a single formulation;
- c) Use of a single molecular entity that combines multiple target actions.
The initial method has the problem of insufficient patient compliance since patients would have to take multiple drugs throughout the day to treat various symptoms. When compared the second and third methods, one key feature is that using a single chemical entity reduces the possibility of drug-drug interactions and enables the advance of novel patentable molecules. However, designing a molecule with several ligands in a suitable ratio is difficult for distinct receptors.
Molecular alteration of known and existing lead nuclei is the current approach in the production of new drugs. Molecular hybridization (MH) is a structural alteration approach that may be used to create novel drugs and prototypes by fusing two or more known bioactive moieties into a single molecule. After the fusing of bioactive subunits recognized by two or more biological receptors, the MH is a technique to build novel prototypes. The MH can help to address the primary reasons for drug discovery failure, such as lack of efficacy and poor safety. The advantage of using MH is that it may activate several sites with a single molecule, enhancing therapeutic efficacy, and also improving bioavailability [71]. The MH method is used to develop the novel ligands for different therapeutic categories, including anti-TB, antithrombotic, cardiovascular, and so on. There are a few instances of heterocyclic hybrids created for TB targets using MH [72].
On the other hand, combining drugs in the same formulation helps pharmaceutical industries to generate new commercial products quickly and extend the patent life of some older treatments. In this type of strategy, investigations on the dominance of combinations and drug-drug interactions are required [73]. Only drug A binds to receptor A and only drug B binds to receptor B. The interaction of drug A with receptor B (and vice versa) is prohibitive, but it is feasible to construct molecules that interact with both receptors and contribute synergistically to the desired effect. Three conditions should be considered while designing a hybrid compound drug:
- a) A spacer agent is used to join the desired subunits;
- b) Both subunits are linked without the use of a spacer agent and fused;
- c) The desired activities are combined into a new structure.
In TB treatment, combining various effects in the same drug is an intriguing technique to improve therapy compliance, increasing activity, and reducing resistance.
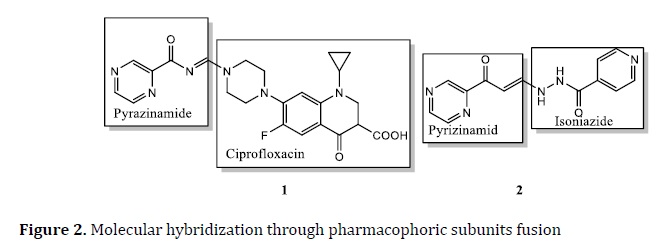
This approach is used in TB drug discovery to increase efficacy and reduce drug resistance. The MH is the scaffold of three anti‒TB drugs: INH, PZ, and ciprofloxacin. The compounds 1 and 2 have good activity against Mtb with MIC=0.78 μg/mL and 0.1 μg/mL, respectively (Figure 2) [74].
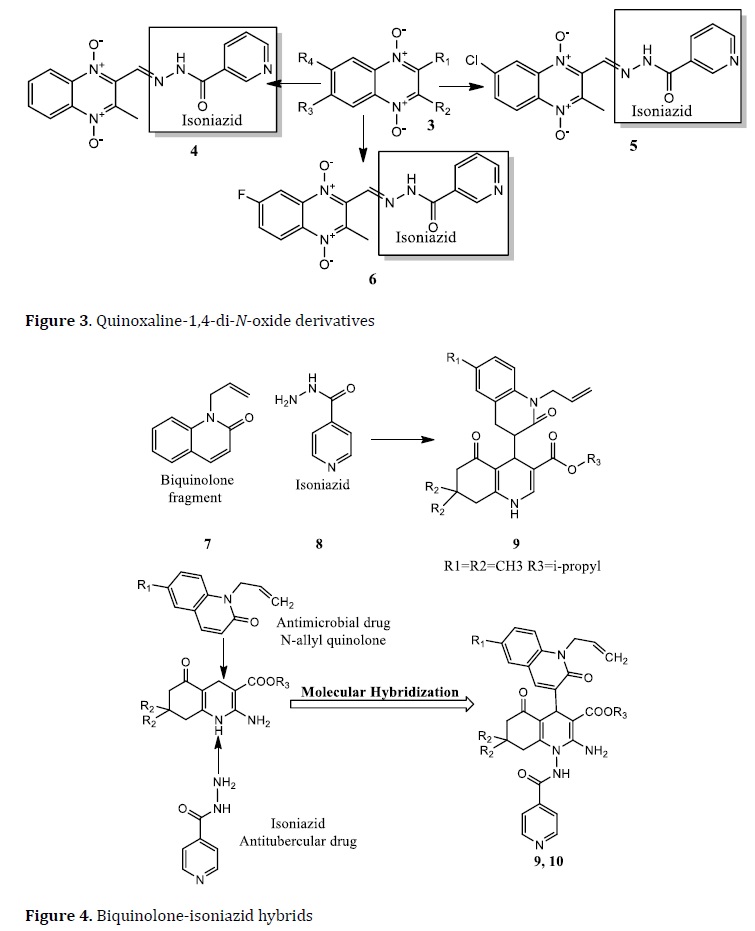
Quinoxaline-1,4-di-N-oxide is used as a new hybrid drug active against Mtb [75]. The anti-TB activity of quinoxaline-1,4-di-N-oxide derivate (3) was obtained by MH with the INH. The halogens in position 7 enhance the anti-TB activity. The compounds with chlorine (4) and fluorine (5) exhibited IC50 values of 1.04 μg/mL and 0.58 μg/mL, respectively. While the hydrogen-substituted compound (6) exhibited IC50 of 1.17 μg/mL (Figure 3) [76]. Another active prototype obtained by MH was developed between the phthalimide subunit in the thalidomide and dapsone. This scaffold was active against M. leprae, but it has interesting activity against M.tb with a MIC value of 3.9 μg/mL [77].
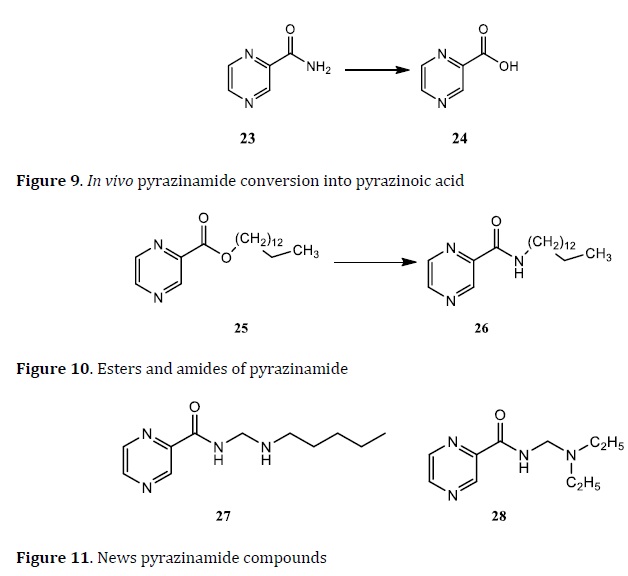
Biquinolone (7) and INH (8) hybrids were designed using the MH technique (Figure 4). Hybrids tested using the brine shrimp bioassay method, compounds 9 and 10 are the most potent compounds with 99% inhibition against Mtb with LC50 values of 35.39 mg/mL and 34.59 mg/mL, respectively [78].
A series of substituted 4H-1,2,4-triazol-3-ylcycloalkanols (Figure 5) was designed by the MH approach, with the essential features of the dehydroquinase (11) and Mtb shikimate kinase enzyme (12) inhibitors. All the hybrids (13-17) exhibited promising activity (MIC 0.59-15.5 μg/mL) against Mtb H37Rv. Compounds 13‒17 exhibited MIC < 1 μg/mL, and CC50 values were non‒toxic to the VERO C1008 cell line, with selectivity indices >28.
A series of quinoxaline 1,4-di-N-oxides (Figure 6) containing INH [76], based on the fusion between INH (19) and a quinoxaline 1,4-di-N-oxide (18), were tested for anti-TB activity against Mtb H37Rv. Among them compounds 20, 21, and 22 showed higher selective indexes >10, with promising anti-TB activity.
This strategy exhibited the use of the MH technique of INH and FQs to increase the anti-TB activity of the compounds (Figure 7). This compound maintains a high survival rate and reduces in vivo the colony-forming unit (CFU) with few lung lesions and reduces splenomegaly [79].

A MH approach was also performed using FQs and PZ via Mannich bases. These compounds were shown in vitro and in vivo anti-TB activity. The compound exhibited higher log P than PZ. The log P is an important parameter to test the compounds due to the lipophilic characteristics of the Mtb wall (Figure 8). In vitro results showed that the compound was more active than PZ against MDR‒TB. The compound was able to reduce the bacterial load in lung and spleen tissues in in-vivo studies [80].
Albert gave the initial definition of the prodrug in 1958, “any molecule that undergoes biotransformation before manifesting its pharmacological effects”. The word latentiation can be used to support this concept. Drug latentiation is defined as "the chemical change of a bioactive substance to generate a new product that will release the parent component following in vivo enzymatic assault." Bioprecursors and carrier prodrugs are the two types of prodrugs. Bioprecursors are a molecular modification approach providing a novel compound for metabolizing enzymes, which then exhibit the biological activity following the biotransformation. This method avoids the carriers’ usage in most cases. Sulindac, acyclovir, and losartan, among others, are examples of drugs that used this method [81]. The labile connection between a carrier group and an active compound is used to create carriers' prodrugs. After chemical or biological biotransformation, this prodrug releases the parental drug and is responsible for the biological action.
In most cases, the prodrug is inert or less active than the parent drug. To produce the synergic effect, the carrier selection might explore two strategies: the first is to use an inactive carrier (nontoxic), and the second is to use active molecules. We may classify the last technique as mutual prodrugs or codrugs. In both cases, the active molecule(s) will be liberated with a kinetic hydrolysis reaction [81]. The drug may have certain flaws in the pharmaceutical, pharmacokinetic, or pharmacodynamic phases. Because the drug is unable to overcome the obstacles as a result of this deficient attribute, the impact is either absent or diminished. This difficulty can be solved by using a prodrug strategy. The drug can work after biotransformation, showing that the impact has been optimized. Various studies used the prodrug concept to identify novel anti-TB drugs [82]. PZ, INH, and ethionamide are examples of anti-TB drugs that might be called bio-precursor prodrugs. Intracellular antimycobacterial pyrazinamidase converts the PZ into pyrazinoic acid. This last one can lower the pH in the Mtb's environment, stopping it from growing. Therefore, pyrazinoic acid may permeate the mycobacterial membrane, reducing cytoplasmic pH and causing membrane transport disruption and energy depletion [55]. The PZ resistance was found in tubercle bacillus with a mutation in the PZ/nicotinamidase gene (pncA).
Pyrazinamide (23) is a prodrug bioconverted by PZs into active form pyrazinoic acid (24) and reduce Mtb growth (Figure 9) [55]. The influence of carriers on PZ (25) and pyrazinoic acid (26), and esters and amides compounds with higher lipophilicity than PZ. The esters derivatives have better activity than amides derivates against Mtb (Figure 10) [83]. A study about PZ carriers led to a series of amino methylene analogs of PZ (27 and 28). These two prototypes (27 and 28) act as anti-TB drugs (Figure 11) [84].
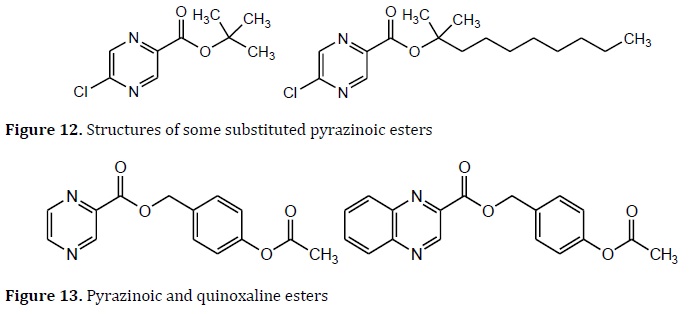
Several pyrazinoic prodrugs with improved lipophilicity are effective against Mtb. Some substituted pyrazinoic esters have 100 times the PZ activity against Mtb and had excellent plasma stability (Figure 12) [85].
Other pyrazinoic (PZ) and quinoxaline esters were tested against Mtb. The compounds 4‒acetoxy‒benzyl esters of pyrazinoic acid and 4'‒acetoxybenzyl 2‒quinoxaline carboxylate showed MIC values 1‒6.25μg/mL (Figure 13) [86].
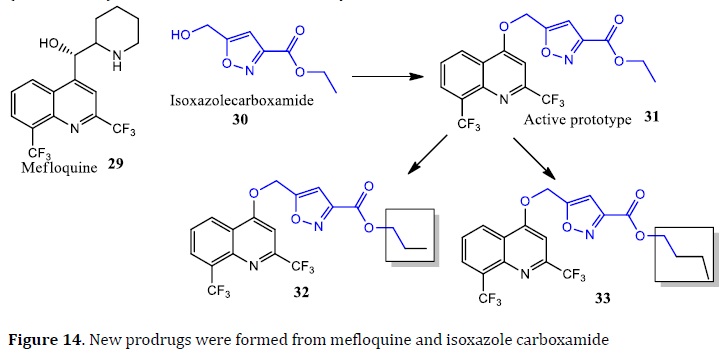
Using the MH, the prodrug 5-(2,8-di(trifluoromethyl)quinolin-4-yloxymethyl)isoxazole-3-carboxylic acid ethyl ester (31) was obtained by the combination of mefloquine fragment (29) and isoxazole carboxamide (30) [87]. Substitutions in prototype 31 to increase carbon chain using propyl and butyl. The compounds 32 were twice less active (IC50 =1.8 μM) than 31 (IC50=0.9 μM), and 33 had similar activity (IC50=0.9 μM) when compared with prototype (Figure 14) [88]. Another study about pyrazinoic (PZ) prodrugs, compared a series of esters and amides prodrugs based on the PZ structure. All compounds exhibited higher lipophilicity (log P) than PZ. The esters derivatives exhibited better in vitro activity against Mtb (MIC=10‒20 μg/mL) than amide derivatives (MIC=> 800 μg/mL).
The prodrug use to create the novel esters of prodrugs like PZ analogs is a key strategy for discovering new drugs with improved pharmacokinetic and pharmacodynamic features. For the Mtb treatment, the INH is a prodrug that is activated by the enzyme katG, which has catalase-peroxidase activity and catalyzes an oxidation process. Following conversion, the drug is bio-transformed into reactive species capable of acylating a mycobacterium enzyme system. The enzyme inhA, which is involved in the formation of mycolic acids, is one of these systems [89]. As a result, after KatG activation, the isonicotinoyl radical combines with NADH, forming an adduct that inhibits Inha. The patient compliance and long-term therapy including many drugs are two of the most difficult aspects of the TB treatment. Mutual prodrugs (or codrugs) are reported to help with therapeutic adhesion issues. The PASA combination in the treatment-and INH was described using the prodrug method. Due to fast phase II metabolism, PASA therapy has several drawbacks, such as gastrointestinal tract (GIT) discomfort and insufficient bioavailability. On the other hand, INH is rapidly absorbed and converted into inert metabolites (acetyl hydrazide, diacetyl hydrazide, nacetylisoniazide, and hydrazine) following the oral treatment [90].
When PAS is applied before this metabolic step (acetylation), the INH half-life is increased. Using the prodrug strategy, it was claimed that combining the two drugs into mutual prodrugs would minimize the GIT toxicity and extensive PASA metabolism, diminish intestinal acetylation of INH, and lengthen the duration of the drug activity (Figure 15) [91]. Polymeric prodrugs were explored to reduce the toxicity and lengthen the INH half-life. To make INH prodrugs, micellar systems of aspartic acid copolymer and N-methylene phosphonic chitosan were used as a carrier. The micellar prodrugs were shown to be effective against Mtb [92].

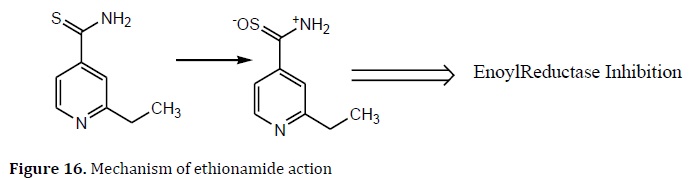
Norfloxacin derivatives were created using the same polymeric prodrugs method. The use of mannosyl ligands as carriers to target macrophages raises drug levels in these cells [93]. Ethionamide is a prodrug that works similarly to INH. After being oxidized by catalase-peroxidase, the medication is bio-converted into ethionamide sulfoxide, an active acylating agent that inhibits inhA enoyl reductase (Figure 16) [94].
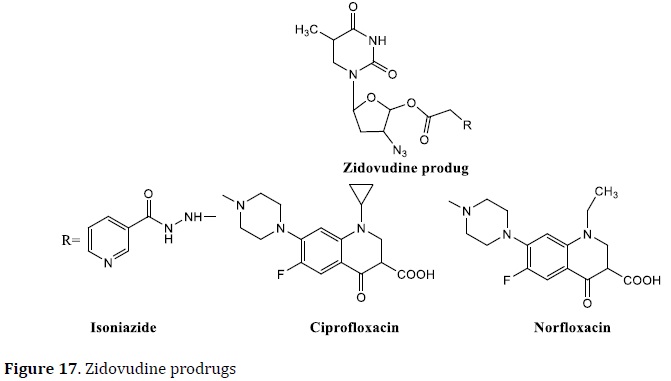
The link between TBs and HIV infection is striking. HIV patients' immune systems are compromised, allowing latent TB to resurface and rendering them more vulnerable to drug-resistant forms. It is expected that two-thirds of people with TB are also HIV positive [79]. The most common HIV therapies combine nucleoside analogs (such as zidovudine) with other drugs. Mutual prodrugs of anti-TB drugs (like INH, norfloxacin, and ciprofloxacin) and HIV nucleoside analogs (zidovudine, stavudine, and lamivudine) were tested in this context. The zidovudine prodrugs were tested at 6.25 μg/mL against Mtb H37Rv and exhibited 99% inhibition (with FQs) and 90% of inhibition (with INH) (Figure 17) [95, 96].
New drug candidates for tuberculosis treatment
Efforts to create anti-TB drugs have risen in the recent years. Despite the advancements, no new drugs have been discovered in the recent several years. The quest for the additional Mtb targets that can be suppressed to remove all known strains is a global effort. Several Mtb targets have been discovered. These targets might be inhibited throughout the growth and latent phases. Some targets in the growth phase are mycolic acid (mycolic acid metabolism), DprE1/DprE2 (cell wall metabolism), MshC (mycothiol ligase), AtpE (ATP synthesis), Def (protein processing), HisG (histidine biosynthesis), GIgE (maltose metabolism), and methionine aminopeptidase (protein processing). Some targets of the dormant phase are isocitrate lyase (energy metabolism), proteasome complex (protein processing), L,D‒transpeptidase (peptidoglycan metabolism), DosR (DevR) (regulation of dormancy), and CarD (stringent response) [97]. The genome sequence of Mtb has driven the largest progress in novel TB target discovery. Nevertheless, the genome-derived target-based strategy has had limited success in the antibacterial class till now [98]. The discovery of new anti-TB drugs should meet certain criteria, such as a broad spectrum against MDR-TB and XDR-TB, an adequate and shorter treatment duration, reducing pill burden, an adequate pharmacokinetic profile to reduce drug-drug interactions (to be administered with HIV drugs), and a long half-life of the drug [99].
Currently, nine drugs are at various phases of clinical trials in the worldwide TB development pipeline. PNU 100480 (protein synthesis inhibitor), AZD 5847 (protein synthesis inhibitor), SQ 109 (cell wall and multitarget inhibitor), OPC67683 (cell wall and multitarget inhibitor), PA824 (cell wall and multitarget inhibitor), gatifloxacin (DNA gyrase inhibitor), moxifloxacin (DNA gyrase inhibitor), TMC 207 (ATP synthase inhibitor), and superb (cell wall and multitarget inhibitor) are among the pipelines. Some of them are active against MDR-TB and XD-TB in both latent and active forms [99]. The majority of current anti-TB drugs under development were created via molecular modification methods. Using bioisosterism as a molecular alteration, the FQs, gatifloxacin, and moxifloxacin were derivatized scaffolds from the parent nalidixic acid (Figure 18). PA824 and OPC67683 were designed via bioisosterism and molecular hybridization on the metronidazole scaffold (Figure 19).
The oxazolidinone derivative PNU‒100480 was designed using linezolid as a parent drug and explored bioisosterism as a molecular modification tool (Figure 20).
The ethambutol was used to develop the compound SQ109 using bioisosterism and molecular hybridization (Figure 21).
New drugs anti-tubercular in a clinical trial
Since the rifampicin discovery in the 1960s, no new drug to treat TB has been put on the market. Several governments and public and private sectors are currently testing drugs in clinical trials [100], like the phase II clinical trial of TMC207and PA‒824 (Figure 22). TMC207 is an ATP synthase inhibitor with a wide anti-TB spectrum. This diarylquinoline showed a high activity against Mtb resistant to INH and PZ with MIC=0.01 and 0.03 μg/mL, respectively [101].
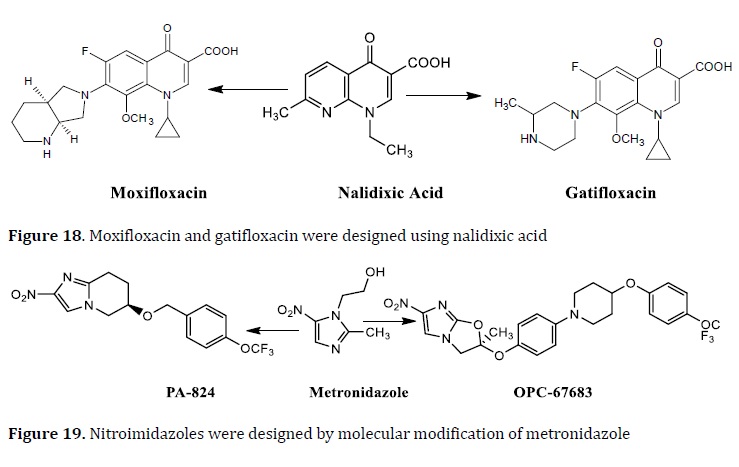

PA-824, a metronidazole analog and is being tested in clinical trials as an anti-TB drug. In vitro activity studies, PA-824 showed a MIC value of 0.015 μg/mL without cross-resistance with the current drugs [102].
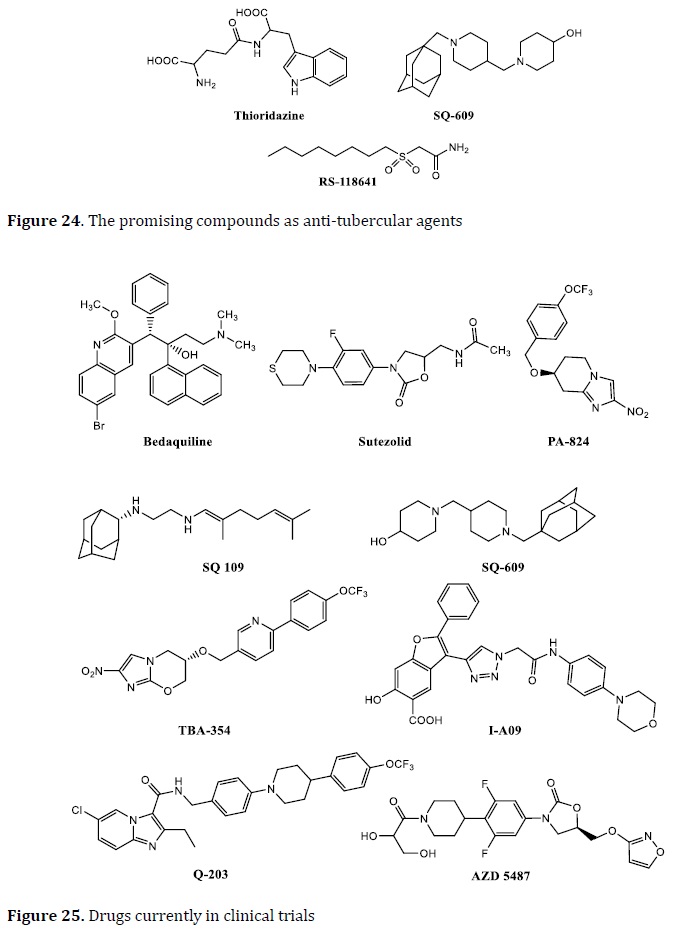
LL3858 is an ethambutol analog called SQ109 (cell wall multitarget inhibitor), and a metronidazole analog called OPC67683 (cell wall multitarget inhibitor) has all just begun clinical trials (Figure 23). Thioridazine, a neuroleptic, has been used as an anti-TB drug that cures XDR-TB patients. SQ609 and RS 118641 are the other two drugs in clinical trials (Figure 24) [100, 103].
Drugs in clinical trials
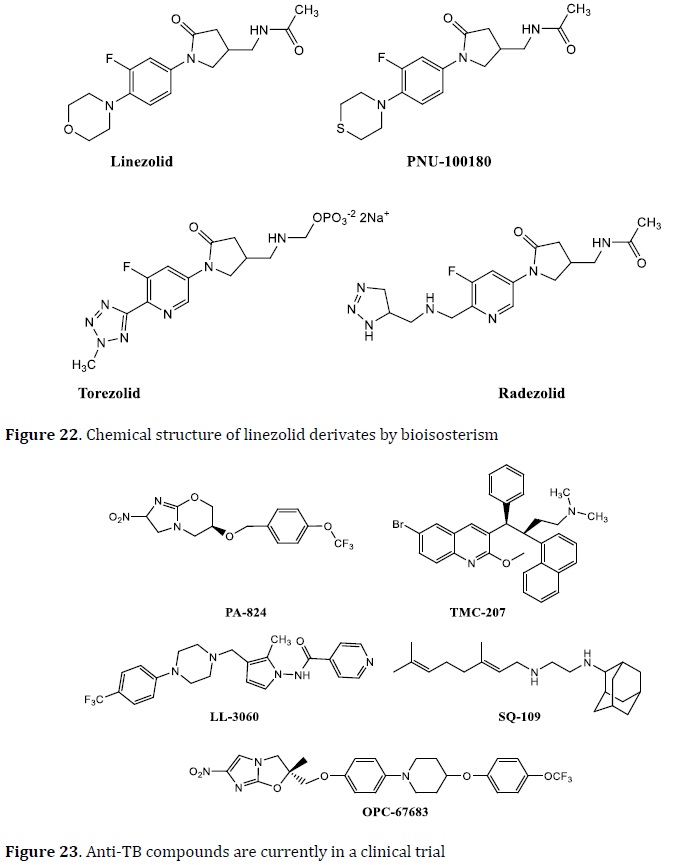
Many anti-TB drugs are in the advanced phases of current clinical trials, and others are in preclinical studies, as anti-tuberculosis research has gained attraction (Figure 25). Some of these drugs, such as bedaquiline, which was authorized by the FDA after 40 years [104], sutezolid, SQ 109, the dipiperidine SQ609 [105], TBA354, IA09, Q 203, PA 824, and AZD 5487 [106] were identified as anti-TB agents and developed as prospective TB treatments.
Bioisosterism
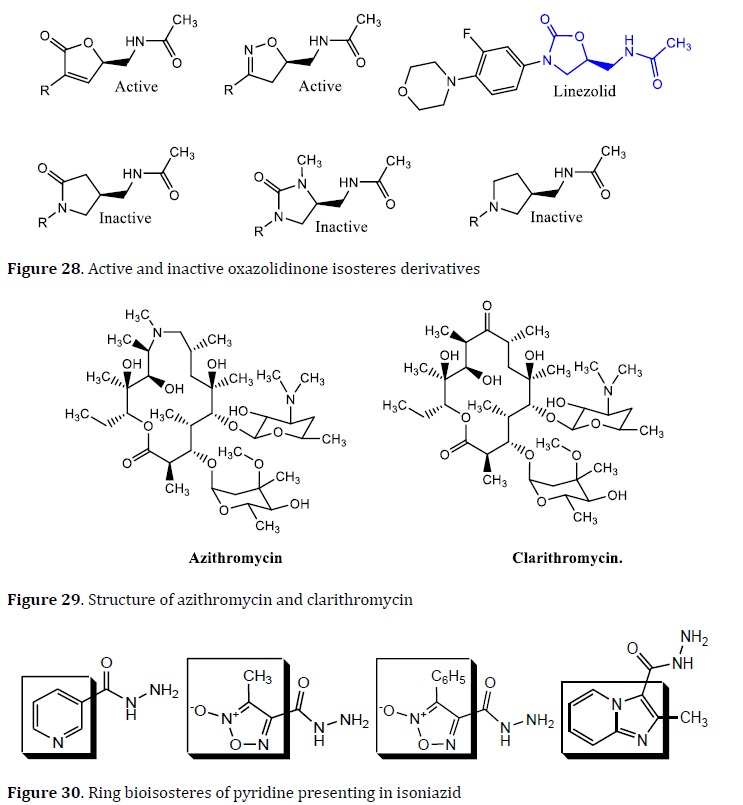
The word "bioisosterism" refers to substances with comparable physicochemical and biological properties. The substitution of molecular fragments, such as a functional group, by another with the equivalent physicochemical qualities is the basis for this molecular modification. The classic and non-classic bioisosterism were separated into two types. Valence atoms, chemical groups, and aromatic rings are used to classify classic bioisosterism (Table 1) [107, 108]. Fluoroquinolones (FQs) are a famous example of bioisosterism-derived medications. Bacterial DNA replication and transcription are inhibited by these substances. The FQs are mostly used in MDR-TB patients. Ciprofloxacin, sparfloxacin, ofloxacin, moxifloxacin, and levofloxacin are the most often utilized FQs in the TB treatment. Linezolid is an antibacterial drug used to treat gram-negative bacterial pneumonia and MDR-TB. As a result, bioisosteres such as PNU100480, radezolid, and torelozid were created [109⎼111].
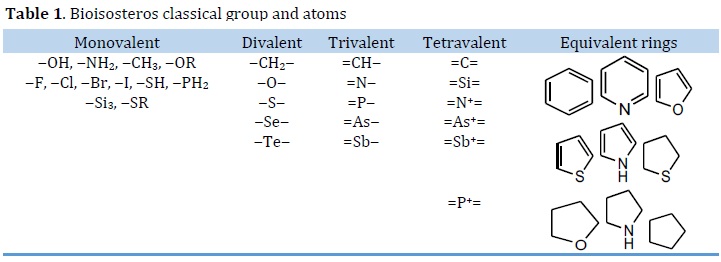
Isosterism describes atoms or organic or inorganic compounds with the same number and/or arrangement of electrons, such as C=O, N=N, CO2, and NO2 [112]. The hydride displacement law, which explains that adding hydrogen to an atom conveys features of the next greatest atomic number (i.e. the fluorine anion (F-) and the hydroxyl anion (HO-) offers some parallels according to Grimm law). Expanding on the idea, isosteres are defined as elements, compounds, or ions that have the same number of electrons at the valence level [113]. Bioisosteres are groupings or compounds that have a chemical and physical resemblance and have biological effects that are generally comparable. The monovalent atoms or groups, divalent atoms or groups, trivalent atoms or groups, tetravalent atoms, and ring equivalents are all split into the traditional bioisosteres (Table 1). Nonclassical bioisosteres lack the steric and electronic definitions of classical isosteres, as well as having the same number of atoms changed in the substituent or moiety. The functional groupings, noncyclic or cyclic, and retroisosterism are examples of nonclassical bioisosteres [114].
FQs, for example, have anti-TB efficacy in addition to Gram-negative and Gram-positive activity. By binding to the DNA gyrase-DNA complex, this family of drugs showed limited bacterial DNA replication and transcription. FQs are mostly used in individuals with MDR-TB. Ciprofloxacin, sparfloxacin, ofloxacin, moxifloxacin, and levofloxacin are the most effective FQs for the TB treatment [115]. The bactericidal efficacy of several FQs against Mtb in the latent and exponential development stages, moxifloxacin and levofloxacin are the most promising drugs [116]. The bioisosteric replacement was used to obtain all of these FQs drugs (Figure 26).
Linezolid is an oxazolidinone drug used in the treatment of nosocomial pneumonia and skin and soft tissue infections caused by Gram-positive bacteria [109]. It exhibited interesting results against MDR‒TB [110]. Some oxazolidinone bioisosteres have been developed. PNU‒100480 is a derivative of linezolid; the other linezolid derivatives such as radezolid and torelozid are formed by isosteric replacement and used in the TB treatment (Figure 27) [111].
Isosteric replacement does not always lead to the equal or more active drugs. Sometimes isosteric replacement leads to inactive drugs (Figure 28). In the bioisosteric approach, it is possible to find at least one (or more) equivalent systems. M. avium-intracellulare complex is one of the general bacterial opportunistic in patients with AIDS.
Clarithromycin and azithromycin are drugs for prevention and treatment. They have bacteriostatic activity due to binding to the 50S ribosomal unit [117] (Figure 29). Bioisosteres of INH was designed to explore the bioisosteric replacement of pyridine ring to imidazo[1,2‒α]‒pyridine. However, 2‒methylimidazo[1,2‒α]pyridine‒3‒carboxylic acid hydrazide displayed less anti-TB activity than INH [118]. After the bioisosteric replacement of pyridine to 1,2,5‒oxadiazole‒2‒oxide (furoxan) (Figure 30).
Drug development for tuberculosis (TB) is now a difficult task. TB therapy is difficult by several issues, together with a lack of efficacy, resistance, poor compliance, and co-morbidities. Molecular alteration is an essential and promising technique to discover novel anti-TB drugs; this fact can be seen in the present pipeline of anti-TB drugs. Furthermore, this technique can answer a swing of issues related to efficacy, treatment length, drug resistance, and side effects. The discovery of novel drugs to treat Mtb infection relies heavily on the molecular alteration. This technique has resulted in the discovery of a more potent and safe drug with broad actions, including against MDR and XDR-TB [119⎼122]. The molecular alteration was used to make the most recent drugs for TB in clinical trials, stressing the relevance of this method in the recognition of the novel anti-TB treatments. The adaptability of molecular hybridization as a structural alteration tool is beneficial in the development of new optimized pharmacophores or prototypes with novel molecular structures. Therefore, it is useful in newly authorized treatments, as well as the emergence of new illnesses and the emergence of MDR strains.
Conclusion
There have been no highly effective drugs discovered to treat TB since the discovery of rifampicin in the 1960s. The development of the novel anti-TB drugs is critical, given the rise in resistance. A new anti-TB drug should have a wide spectrum of action, adequate posology to ensure patient compliance, a short-term treatment, and satisfactory pharmacokinetic properties. The molecular alteration approach has shown to be one of the most promising approaches to bring a novel drug to market. This method has been used to produce some drugs currently on the market. To design and develop novel drugs against Mtb, molecular alteration procedures such as molecular hybridization, prodrug approach, and bio-isoterism have been used. The current therapy has several drawbacks, like side effects, Mtb resistance, and comorbidity. The BCG (Bacillus Calmette-Guérin) vaccination, which is an attenuated M. Bovis, has been used, although its success is debatable. All of these MDR-related issues motivate the development of the novel, safer, and more effective drugs with fewer adverse side effects.
Acknowledgments
The authors are grateful to Glocal School of Pharmacy, Glocal University, Saharanpur, Uttar Pradesh, India, and Laboratory Medicine Department, Faculty of Applied Medical Sciences, Umm Al‒Qura University, Makkah, Saudi Arabia for providing the necessary facilities, and support to carry out this work.
Conflict of Interest
The authors declare that there is no conflict of interest.
The authors’ contributions
Saad Alghamdi and Mohammad Asif have designed and prepared the manuscript and both are contributed to the preparation and revision of the manuscript.
Orcid
Saad Alghamdi 0000‒0003‒4532‒9128
Mohammad Asif 0000‒0002‒9352‒3462
How to cite this manuscript: Antitubercular drugs: new drugs designed by molecular modifications. Asian Journal of Green Chemistry, 6(3) 2022, 327-354. DOI: 10.22034/ajgc.2022.4.4
)